Seurat空间转录组分析(一)数据读入

关于空间转录组分析的学习,我推荐先学习单细胞转录组分析,熟练掌握单细胞的数据读入,常规分析,整合去批次,以及部分高级分析(例如拟时序、转录因子和细胞通讯分析),在这个基础上,理解和学习单细胞空间转录组就非常快了,Seurat官方文档(https://satijalab.org/seurat/articles/spatial_vignette.html)就是一个很好的入门教程。
在学习此空转教程之前,我先介绍一下空转数据如何读入R语言,然后构建成Seurat对象。
一. 导读
空间数据如何储存在Seurat中?
来自10x的visium数据包括以下数据类型:(如果是其它空间单细胞产品,后面再慢慢聊)
- 通过基因表达矩阵得到一个点(spot )
- 组织切片图像(采集数据时H&E染色)
- 用于显示的原始高分辨率图像与低分辨率图像之间的比例因子。
在Seurat对象中,Spot by基因表达矩阵与典型的“RNA”分析类似,但包含spot水平,而不是单细胞水平的数据。图像本身存储在Seurat对象中的一个images 槽(slot)中。图像槽还存储必要的信息,以将斑点与其在组织图像上的物理位置相关联。
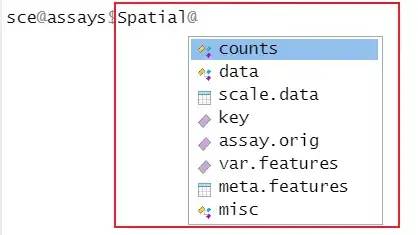
image-20230312105819088
空转下游和单细胞类似的处理,主要包括:
- Cellrange下机,读入R为Seurat对象;
- 双细胞预测(可选);
- 低质量的细胞过滤(可选);
- 标准化特征选择和归一化;
- 降维聚类;
- 分群注释;
- 差异表达分析得到特征markers。
从注释完的数据其,其后的分析一般可以归为个性化分析,和单细胞分析一样,主要有:
- 富集分析:如GO、KEGG和GSEA富集分析;
- 转录因子分析;
- 空转拟时序分析;
- 空转细胞通讯分析;
- ......
二. 空转数据如何读入R语言
Step1. R包加载及安装
library(Seurat)
library(SeuratData)
library(ggplot2)
library(patchwork)
library(dplyr)
Step2. 加载数据
针对不同的数据类型有不同的加载策略:
(1)加载Seurat官网的示例数据
示例数据在https://support.10xgenomics.com/spatial-gene-expression/datasets获得,并使用Load10X_Spatial()函数将其加载到Seurat。这将读取spaceranger管道的输出,并返回Seurat对象,该对象包含spot级别的表达数据以及相关的组织切片图像。
本文的示例数据是10X平台的,内置于SeuratDataR包,加载方式如下:
InstallData("stxBrain")
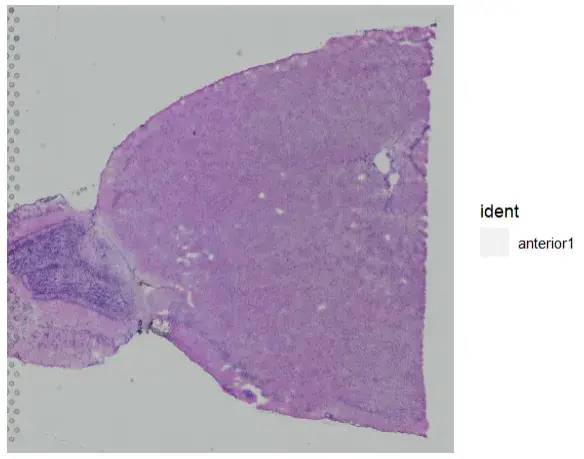
brain <- LoadData("stxBrain", type = "anterior1")
brain
# An object of class Seurat
# 31053 features across 2696 samples within 1 assay
# Active assay: Spatial (31053 features, 0 variable features)
## 检查示例数据
head(brain@meta.data)
# orig.ident nCount_Spatial nFeature_Spatial slice region
# AAACAAGTATCTCCCA-1 anterior1 13069 4242 1 anterior
# AAACACCAATAACTGC-1 anterior1 37448 7860 1 anterior
# AAACAGAGCGACTCCT-1 anterior1 28475 6332 1 anterior
# AAACAGCTTTCAGAAG-1 anterior1 39718 7957 1 anterior
# AAACAGGGTCTATATT-1 anterior1 33392 7791 1 anterior
# AAACATGGTGAGAGGA-1 anterior1 20955 6291 1 anterior
SpatialDimPlot(brain,alpha = 0)
#更多参数参考
?SpatialDimPlot()
(2)加载10X Cellrange上游输出的数据
常规流程是不会使用LoadData函数进行读取数据,因为正常情况下我们拿到的是10 X Space Ranger的输出结果:
以下是下载自10X官方网站的数据:
https://www.10xgenomics.com/resources/datasets?query=&page=1&configure%5Bfacets%5D%5B0%5D=chemistryVersionAndThroughput&configure%5Bfacets%5D%5B1%5D=pipeline.version&configure%5BhitsPerPage%5D=500&configure%5BmaxValuesPerFacet%5D=1000&menu%5Bproducts.name%5D=Spatial%20Gene%20Expression
test_data = Load10X_Spatial(data.dir = "./input/",
filename = "Visium_FFPE_Human_Normal_Prostate_filtered_feature_bc_matrix.h5",
assay = "Spatial",
slice = "test")
head(test_data@meta.data)
SpatialDimPlot(test_data,alpha = 0)
#和单细胞数据一样,可以人为更改这些命名
test_data@project.name <- "test"
Idents(test_data) <- "test"
test_data$orig.ident <- "test"
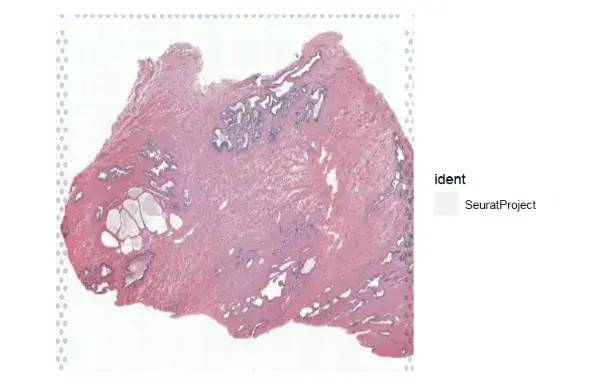
image-20230224173610241
## 获取切片图像坐标:
img<- GetTissueCoordinates(test_data)
img$imagerow = 540-img$imagerow
write.csv(img,file = "./No_IHC/position_information.csv")
head(img)
# imagerow imagecol
# AAACAAGTATCTCCCA-1 183.3725 434.0820
# AAACAATCTACTAGCA-1 442.7995 247.5663
# AAACAGAGCGACTCCT-1 381.8799 409.0720
# AAACAGCTTTCAGAAG-1 222.4493 139.4663
# AAACCCGAACGAAATC-1 210.8508 475.3213
# AAACCGGAAATGTTAA-1 161.2021 503.7606
(3)非常规数据的读入
3.1 缺少IHC图像
有些时候从数据库中下载得到的数据,由于缺少IHC图像,可以利用以下方式进行读取:
# 把空间数据当成单细胞数据读入
test_data2 = Read10X("./input/filtered_feature_bc_matrix/")
test_data2 <- CreateSeuratObject(counts = test_data2,
min.features = 0,
project = "test")
test_data2
# An object of class Seurat
# 17943 features across 2543 samples within 1 assay
没有IHC的空转数据,作者一般会提供一个position information:
# 读入单细胞的位置信息
position = read.csv("./No_IHC/position_information.csv",header = T,row.names = 1)
head(position)
position = select(position,imagecol,imagerow)
#将位置信息合并入单细胞seurat对象
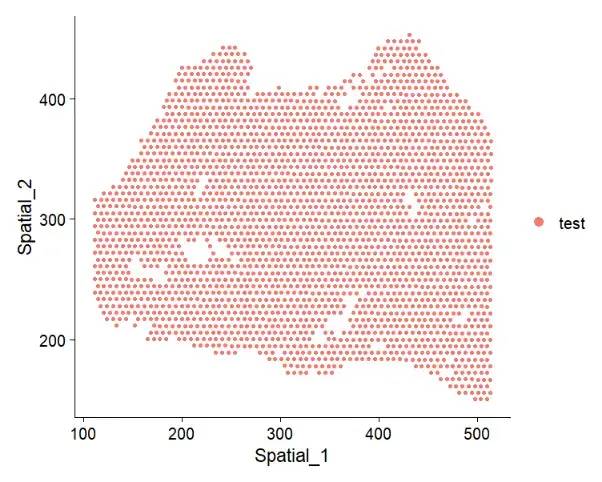
colnames(position) = paste0("Spatial_",1:ncol(position))
test_data2[["spatial"]] <- CreateDimReducObject(embeddings = as.matrix(position),
key = "Spatial",assay = "RNA")
DimPlot(test_data2,reduction = "spatial",pt.size = 1)
3.2 缺少IHC图像也可采用Slide-seq的方法
test_data2 = Read10X("./input/filtered_feature_bc_matrix/")
test_data2 <- CreateSeuratObject(counts = test_data2,
min.features = 0,
project = "test",
assay="Spatial")
test_data2
# An object of class Seurat
# 17943 features across 2543 samples within 1 assay
# Active assay: RNA (17943 features, 0 variable features)
coord.df = read.csv("./position_information.csv",header = T,row.names = 1)
test_data2@images$image = new(
Class = 'SlideSeq',
assay = "Spatial",
key = "image_",
coordinates = coord.df
)
test_data2
SpatialDimPlot(test_data2)
以上就是空转数据读入R语言的常见的几种方法。
- END -