课前准备--单细胞突变矩阵的获得与有害位点的识别
原创课前准备--单细胞突变矩阵的获得与有害位点的识别
原创
追风少年i
发布于 2024-06-18 11:42:30
发布于 2024-06-18 11:42:30
作者,Evil Genius
22号马上上课了,该准备的基本都准备完毕,静待上课就可以了。当然期间还会根据情况进行更新
单细胞测序数据生成的bam文件,经过cellsnp-lite分析时候拿到如下的文件:
关于这个cellsnp-lite,我发现很多人的用法都是错误的,这个会在课上详细说一下该怎么用。
首先是call snp 的文件
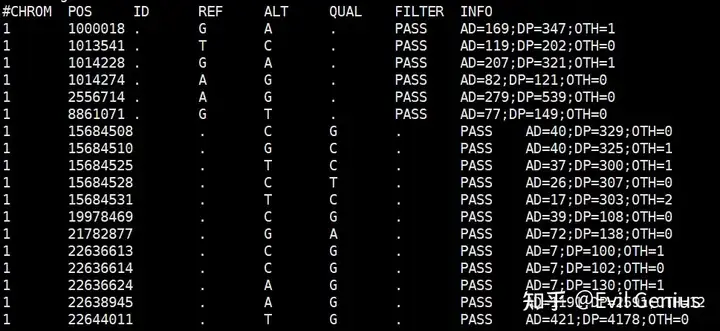
大家注意这个文件,还是根据基因组的位点找到的突变信息,并没有注释到具体的基因,以及是否引起氨基酸的变化,所以我们需要注释一下,拿到如下的结果
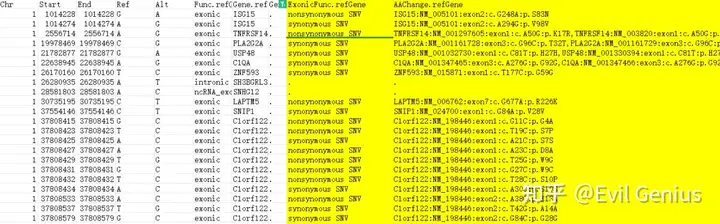
这个时候就拿到突变的氨基酸变化信息,至于变化时候有害,需要额外的注释,或者数据库查找,这个之前分享过,下图是示例:
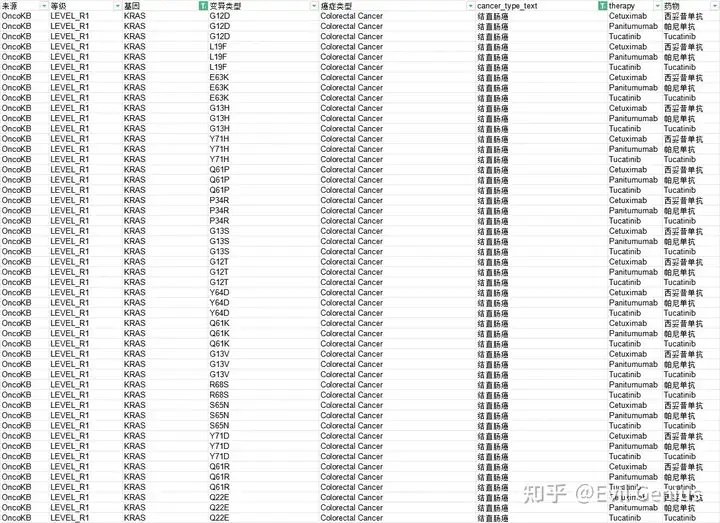
大家可以查阅,比如clinvar、oncokb、my cancer genome等数据库。
接下来就是要拿到单细胞的突变矩阵,如下图:
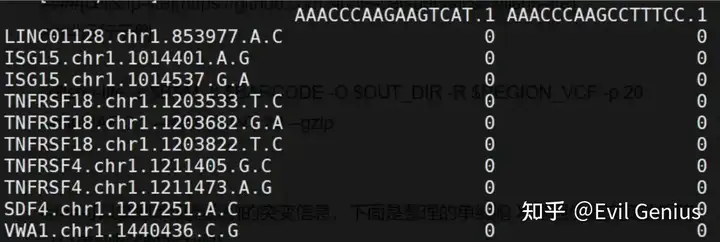
拿到完整的信息之后,就可以跟文章一样纳入单细胞的基础分析之中了。
好了,拿到cellsnp-lite的分析结果,我们首先来注释位点的氨基酸变化,软件是ANNOVAR,做过外显子的应该都很熟悉的
table_annovar=table_annovar.pl脚本路径 humandb=humandb数据库路径
perl $table_annovar \
--buildver hg38 \
--otherinfo \
--nastring . cellSNP.base.vcf $humandb \
-protocol refGene \
-operation g \
--vcfinput --remove > test.log 2>&1 即可得到单细胞突变的注释文件
接下来是矩阵的整理
import scipy.io as sio
matrix_data = sio.mmread('cellSNP.tag.AD.mtx')
matrix_data = pd.DataFrame(matrix_data.todense())
###barocde
barcode = pd.read_csv('cellSNP.samples.tsv',sep = '\t',header=None)
matrix_data.columns = barcode.iloc[:,0] 处理注释文件
anno = pd.read_csv('cellSNP.base.vcf.hg38_multianno.txt',sep = '\t')
anno['index'] = anno['Gene.refGene'] + '.' + anno['Start'] + '.' + str(anno['Ref']) + '.' + str(anno['Alt'])
matrix_data.index = anno['index'] matrix_data.to_csv('single.snp.xls',sep = '\t') 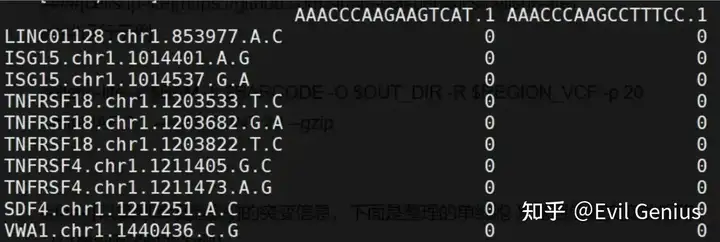
生活很好,有你更好
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者
