CellRanger ARC—单细胞RNAseq和ATAC联合分析套件

CellRanger ARC—单细胞RNAseq和ATAC联合分析套件

工欲善其事必先利其器
Cell Ranger ARC
CellRanger ARC 是10x Genomics 专为单细胞多组学数据分析设计,同时分析单细胞RNA测序(scRNA-seq)和单细胞ATAC测序(scATAC-seq)数据的一组分析pipline 。
主要包含以下几个方面:
- 数据预处理
- 将原始的测序数据转换为可分析的数据格式。
- 包括对FASTQ文件的读取、质量控制、去重复和对齐等步骤。
- 细胞检测和定量
- 自动检测单个细胞,并对每个细胞进行转录组和染色质可及性数据的定量分析。
- 生成包含基因表达和染色质状态的矩阵文件。
- 多组学整合
- 将scRNA-seq和scATAC-seq数据整合在一起,进行联合分析。
- 可以揭示基因表达和染色质可及性之间的关系。
- 可视化和下游分析
- 提供多种数据可视化选项,如UMAP或t-SNE图,用于展示细胞的聚类和特征。
- 支持下游的生物信息学分析,如差异表达分析、轨迹分析等。
官网:
- https://support.10xgenomics.com/single-cell-multiome-atac-gex/software/overview/welcome
如何安装
10X的软件安装都很简单,官网下载解压即可使用。
官网下载链接:
- https://support.10xgenomics.com/single-cell-multiome-atac-gex/software/downloads/latest
首先是需要注册一下信息
申请
填写信息后,可跳转到下载界面
下载软件及所需的参考文件
## 软件下载
wget -O cellranger-arc-2.0.2.tar.gz "https://cf.10xgenomics.com/releases/cell-arc/cellranger-arc-2.0.2.tar.gz?Expires=1721143948&Policy=eyJTdGF0ZW1lbnQiOlt7IlJlc291cmNlIjoiaHR0cHM6Ly9jZi4xMHhnZW5vbWljcy5jb20vcmVsZWFzZXMvY2VsbC1hcmMvY2VsbHJhbmdlci1hcmMtMi4wLjIudGFyLmd6IiwiQ29uZGl0aW9uIjp7IkRhdGVMZXNzVGhhbiI6eyJBV1M6RXBvY2hUaW1lIjoxNzIxMTQzOTQ4fX19XX0_&Signature=Lx8Sg8mwwlYvUtxVwNl0LmxwtSEVpZy~W6XO5Y~1c~4O7P8zyzNcijN0H~FzE4P10CTc~btSMa5RpaNQN~AqLed~CxDikYWvuMoWb9ffMm3dxu4i1M31aUQtI4DzbGpeOSKl4--VL6qpDNyrSGwiS-T-72C14pdEKEur8m1RPNq8pfxHGc-5eRvvyliJrDN49zJRR1Dnr0LH5valgNTCqzCio5-UApkdl9JLNa07-kvKkh4T7hUecoEpoDGzugOuejJGU1kJzmZoOlQVqwoGUepeQUG6wrqD0~CYj19-tyPXxWCUKPjVZBlk8ERY2trWcr2Z6QB4qqAui~EJguWtwg__&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA"
## 软件大小:
699M 7月 10 23:26 cellranger-arc-2.0.2.tar.gz
## 解压即可使用
tar -xf cellranger-arc-2.0.2.tar.gz
##人参考基因组下载
wget -c https://cf.10xgenomics.com/supp/cell-arc/refdata-cellranger-arc-GRCh38-2020-A-2.0.0.tar.gz
##文件大小
14G 5月 3 2021 refdata-cellranger-arc-GRCh38-2020-A-2.0.0.tar.gz
##解压后21G
21G refdata-cellranger-arc-GRCh38-2020-A-2.0.0
##小鼠参考基因组下载
wget -c https://cf.10xgenomics.com/supp/cell-arc/refdata-cellranger-arc-mm10-2020-A-2.0.0.tar.gz
##文件大小
13G 6月 21 01:04 refdata-cellranger-arc-mm10-2020-A-2.0.0.tar.gz
##解压后19G
19G refdata-cellranger-arc-mm10-2020-A-2.0.0
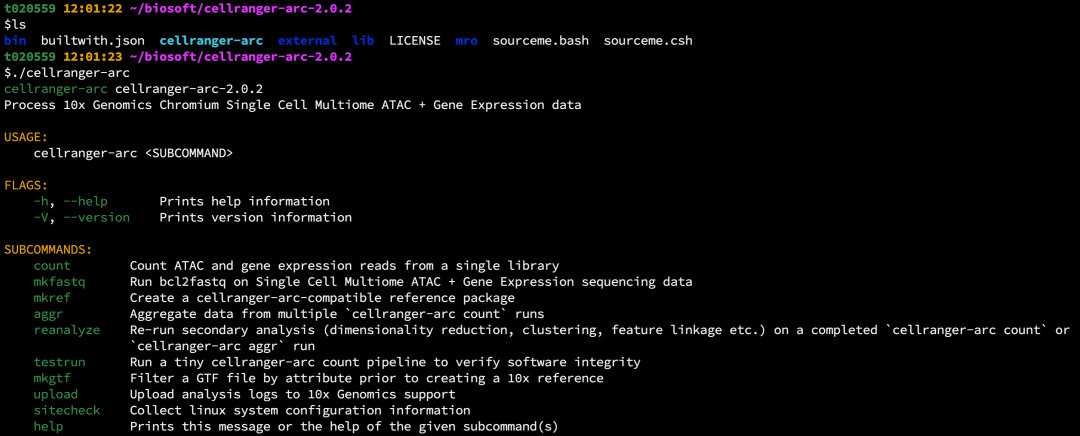
如果需要自定义参考基因组文件,可参考:
- https://support.10xgenomics.com/single-cell-multiome-atac-gex/software/pipelines/latest/advanced/references
功能简述
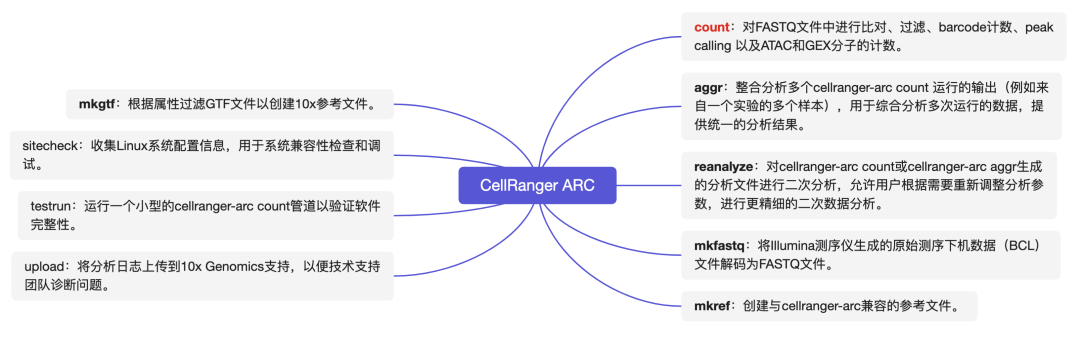
功能模块
最小化使用
通常我们用到的是count定量
cellranger-arc count --id=sampleid \
--reference=/opt/refdata-cellranger-arc-GRCh38-2020-A-2.0.0 \
--libraries=/home/jdoe/runs/libraries.csv \
--localcores=16 \
--localmem=64
--id #运行ID符,同时也是pipline的输出结果目录。可以使用字母、数字、下划线和连字符任意定义,保证唯一即可且不超64个字符即可,一般使用样本名来作为ID
--reference #参考基因组文件
--libraries # 一个3列的CSV文件,文件声明了输入ATAC和GEX FASTQ的fastq文件路径、样本名和库类型
--localcores #指定调用线程数,默认调用系统所有可用内核
--localmem #指定调用的内存大小 ,默认调用系统90%的可用内存
需要特别注意的是CSV输入文件的格式,如果格式有误,程序则无法运行
文件包含3列
- fastq文件路径
- 样本名:fastq文件的前缀
- 数据类型:其中,RNAseq数据是
Gene ExpressionATAC数据是Chromatin Accessibility
fastqs,sample,library_type #固定格式,不能修改
/home/jdoe/runs/HNGEXSQXXX/outs/fastq_path,example,Gene Expression #按需修改
/home/jdoe/runs/HNATACSQXX/outs/fastq_path,example,Chromatin Accessibility #按需修改
一个实例
了解了基本用法,我们来运行一个实例数据看看。使用的数据来自于一项关于人类免疫缺陷病毒(HIV)潜伏期逆转的单细胞多组学分析研究,研究者采用了scRNA-seq和scATAC-seq方法,同时分析了约125,000个经过三种不同潜伏期逆转剂(LRAs)激活的潜伏感染的初级CD4+ T细胞的转录组和表观基因组特征。研究题目是 “Integrated Single-cell Multiomic Analysis of HIV Latency Reversal Reveals Novel Regulators of Viral Reactivation” 。数据已上传GEO数据库,编号 GSE210146 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE210146)。
下载数据和拆分
数据集我们从ENA数据库下载:
- PRJNA864047: https://www.ebi.ac.uk/ena/browser/view/PRJNA864047
cat filereport_read_run_PRJNA864047_tsv.txt |awk -F "\t" -v OFS="\t" 'NR>1{print $3,$8,$17,$19}' |grep "DMSO"|cut -f 3 > down_ascp.txt
## ascp 下载
cat ../a_spinfo/down_ascp.txt |while read id;do(ascp -QT -l 100m -P33001 -i ~/miniconda3/envs/kingfisher/etc/asperaweb_id_dsa.openssh era-fasp@${id} ./);done 1>ascp.log 2>&1
下载文件大小:
$cat down_ascp.txt
fasp.sra.ebi.ac.uk:/vol1/srr/SRR207/010/SRR20724710/SRR20724710.lite
fasp.sra.ebi.ac.uk:/vol1/srr/SRR207/002/SRR20724702/SRR20724702.lite
fasp.sra.ebi.ac.uk:/vol1/srr/SRR207/001/SRR20724701/SRR20724701.lite
fasp.sra.ebi.ac.uk:/vol1/srr/SRR207/009/SRR20724709/SRR20724709.lite
3.8G 7月 11 00:29 SRR20724710.lite
3.4G 7月 11 01:22 SRR20724702.lite
3.4G 7月 11 01:45 SRR20724701.lite
3.5G 7月 11 12:01 SRR20724709.lite
拆分数据
ls SRR*.lite | while read id;do(fasterq-dump -O ~/scRNA/PRJNA692722_atac/b_rawdata/fq_data/ --split-files -e 10 --include-technical ${id} && pigz -p 10 ~/scRNA/PRJNA692722_atac/b_rawdata/fq_data/${id}_*.fastq);done 1>split.log 2>&1

拆分后文件
可以看到每个SRA文件都拆分出来4个fastq文件
文件重命名使其符合cellranger-arc 输入要求
这一步也需要特别注意一下,文件命名不要搞错。
首先我们要知道的是cellranger-arc的文件命名规则
fastq文件命名同10X cellranger的格式要求即:[Sample Name]_S[Number]_L00[Lane Number]_[Read Type]_001.fastq.gz
GEX FASTQs
对应RNAseq数据,我们已经熟知,对于 I1、I2、R1、R2 四个文件,我们只需要确定R1 ,R2 作为输入即可正常运行
I1: Dual index i7 read (optional)I2: Dual index i5 read (optional)R1: Read 1R2: Read 2
ATAC FASTQs
有区别的一点是ATAC的数据,其需要输入的4个文件对应的命名为:
I1: Dual index i7 read (optional)R1: Read 1R2: Dual index i5 readR3: Read 2
或者
I1: Dual index i7 read (optional)R1: Read 1I2: Dual index i5 readR2: Read 2
这时候在文件名重名的时候就要特别区分,我们拆分出来的1,2,3,4四个文件,分别对应的是哪个。
重命名
这里贴一下重命名的脚本。由于示例所用的RNAseq的数据和ATAC数据命名规则还不一致,所以我们需要修改这个脚本分别来命名。
对RNAseq数据重命名
$cat rename_rna.sh
#!/bin/bash
#
rn_file=${1}
out_dir=${2}
declare -A sample_map
declare -A sample_count
declare -A srr_sample_number_map
while read -r key value; do
sample_map["$key"]="$value"
done < ${rn_file}
for file in *.fastq.gz; do
if [[ $file =~ (SRR[0-9]+)\.lite_([0-9]+)\.fastq\.gz ]]; then
original_number="${BASH_REMATCH[1]}"
read_number="${BASH_REMATCH[2]}"
sample_name="${sample_map[$original_number]}"
if [[ -n "$sample_name" ]]; then
if [[ -z "${srr_sample_number_map[$original_number]}" ]]; then
if [[ -z "${sample_count[$sample_name]}" ]]; then
sample_count["$sample_name"]=0
fi
sample_count["$sample_name"]=$((sample_count["$sample_name"] + 1))
srr_sample_number_map["$original_number"]=${sample_count["$sample_name"]}
fi
sample_number=${srr_sample_number_map["$original_number"]}
case $read_number in
1)
new_read_number="R1"
;;
2)
new_read_number="R2"
;;
*)
echo "Unexpected read number: $read_number"
continue
;;
esac
new_file_name="${sample_name}_S${sample_number}_L001_${new_read_number}_001.fastq.gz"
ln -s ${PWD}/"$file" ${out_dir}/"$new_file_name"
echo "Renamed $file to $new_file_name"
fi
fi
done
对ATAC数据重命名
#!/bin/bash
#
rn_file=${1}
out_dir=${2}
declare -A sample_map
declare -A sample_count
declare -A srr_sample_number_map
while read -r key value; do
sample_map["$key"]="$value"
done < ${rn_file}
for file in *.fastq.gz; do
if [[ $file =~ (SRR[0-9]+)\.lite_([0-9]+)\.fastq\.gz ]]; then
original_number="${BASH_REMATCH[1]}"
read_number="${BASH_REMATCH[2]}"
sample_name="${sample_map[$original_number]}"
if [[ -n "$sample_name" ]]; then
if [[ -z "${srr_sample_number_map[$original_number]}" ]]; then
if [[ -z "${sample_count[$sample_name]}" ]]; then
sample_count["$sample_name"]=0
fi
sample_count["$sample_name"]=$((sample_count["$sample_name"] + 1))
srr_sample_number_map["$original_number"]=${sample_count["$sample_name"]}
fi
sample_number=${srr_sample_number_map["$original_number"]}
case $read_number in
1)
new_read_number="R1"
;;
2)
new_read_number="R2"
;;
3)
new_read_number="R3"
;;
4)
new_read_number="I1"
;;
*)
echo "Unexpected read number: $read_number"
continue
;;
esac
new_file_name="${sample_name}_S${sample_number}_L001_${new_read_number}_001.fastq.gz"
ln -s ${PWD}/"$file" ${out_dir}/"$new_file_name"
echo "Renamed $file to $new_file_name"
fi
fi
done
注意:该重命名脚本使用需完全理解,然后按自己数据实际情况修改,以免改错
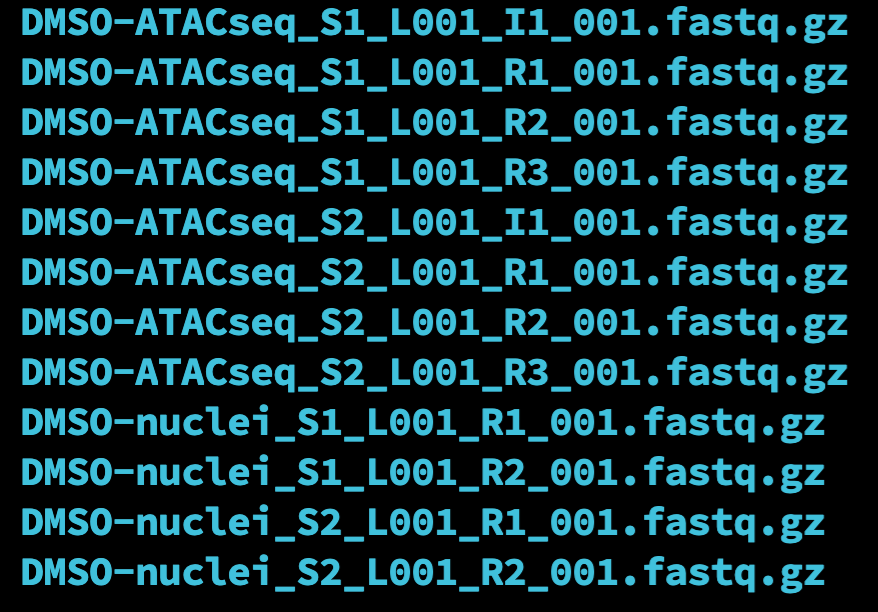
重命名后的文件
构建如下所示的CSV文件
$cat DMSO.csv
fastqs,sample,library_type
/home/data/t020559/scRNA/PRJNA692722_atac/b_rawdata/fq_data/,DMSO-nuclei,Gene Expression
/home/data/t020559/scRNA/PRJNA692722_atac/b_rawdata/fq_data/,DMSO-ATACseq,Chromatin Accessibility
cellranger arc 定量
准备好以上工作就可以提交任务,运行定量流程了
##定量脚本 arc_homo.sh
#! /bin/bash -xe
#
bin=/home/data/t020559/biosoft/cellranger-arc-2.0.2/cellranger-arc
db=/home/data/t020559/ref/homo/refdata-cellranger-arc-GRCh38-2020-A-2.0.0
id=${1}
csv=${2}
/usr/bin/time -v ${bin} count --id=${1} \
--reference=${db} \
--libraries=${2} \
--localcores=8 \
--localmem=100
提交任务
nohup bash /home/data/t020559/scRNA/arc_homo.sh DMSO DMSO.csv 1>log_DMSO.txt 2>&1 &
结果文件
定量后,我们需要的结果文件主要在/path/DMSO/outs 目录下
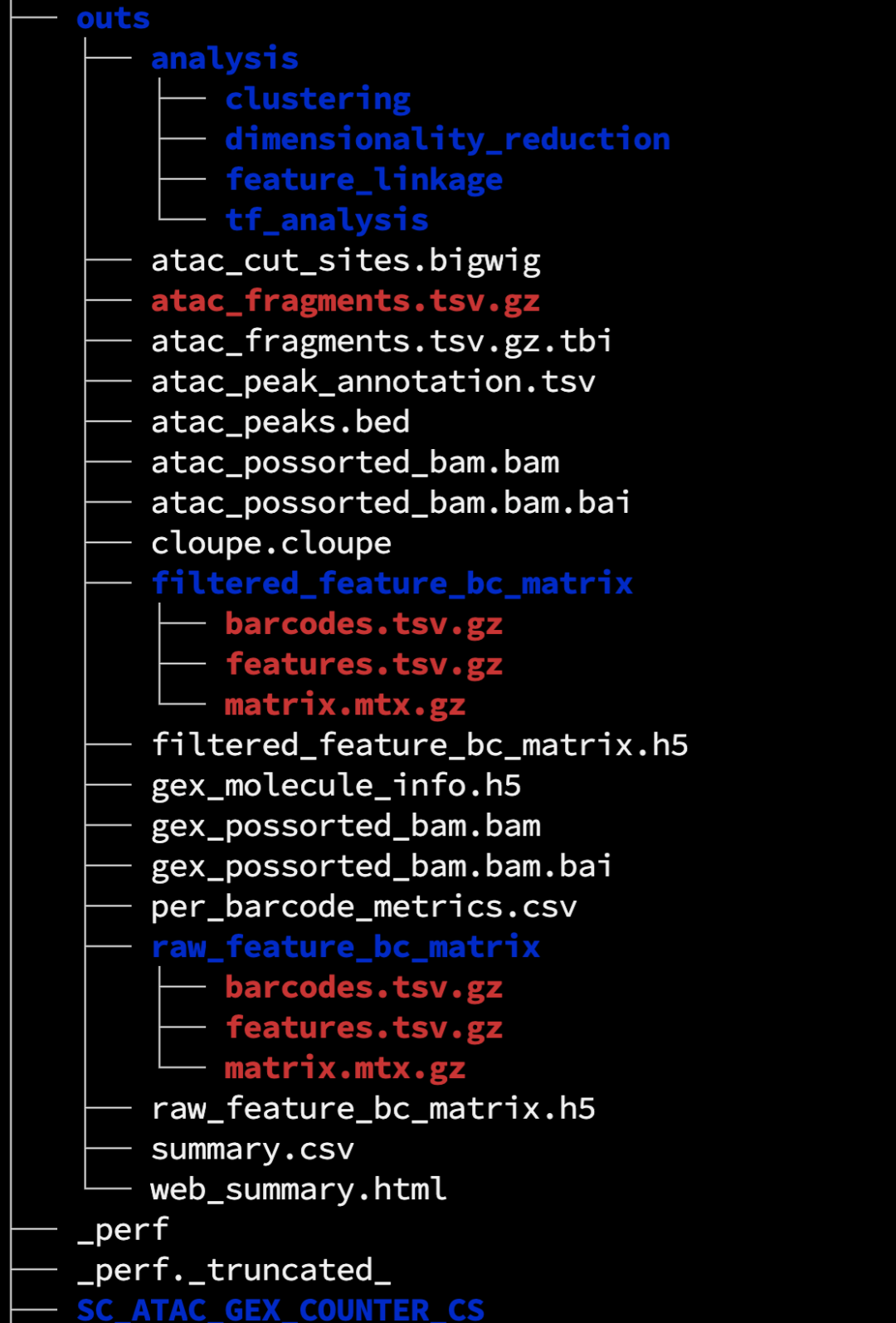
结果文件
web_summary.html结果报告的html文件。raw_feature_bc_matrix.h5原始的cell × features矩阵的h5格式结果文件。行包括了peaks和基因在内,列为每个细胞的barcodes。raw_feature_bc_matrix原始的cell × features矩阵的结果目录,里面就是三个我们熟知的10X标准结果文件(barcodes、features、matrix),其中features包括了peaks和基因在内,且有5列。atac_peaks.bedPeak Calling的结果。atac_peak_annotation.tsvPeak临近基因注释结果
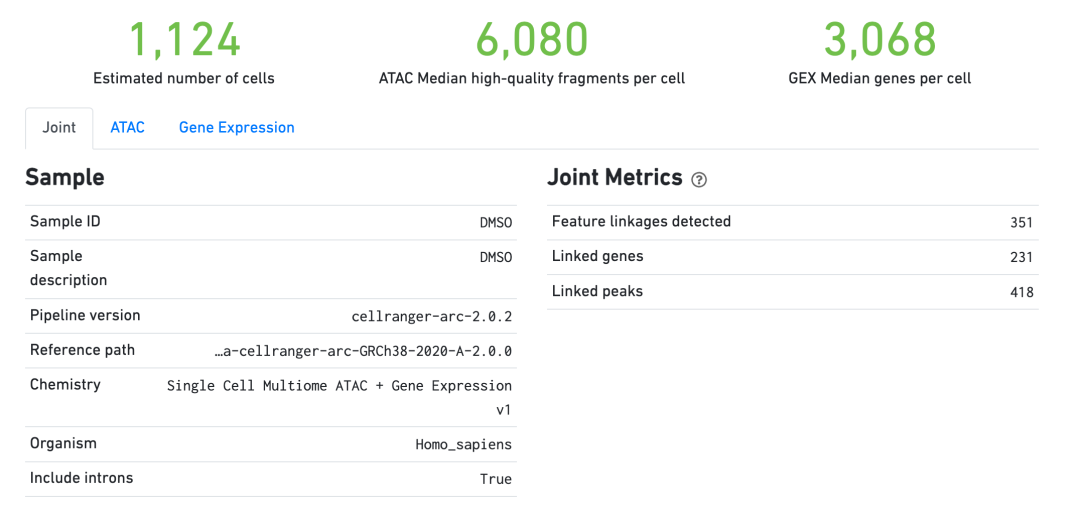
web_summary.html
参考:
- https://support.10xgenomics.com/single-cell-multiome-atac-gex/software/pipelines/latest/using/count#libraries
- https://mp.weixin.qq.com/s/M9E-tGPKlSH0CUPlS_LE2A
腾讯云开发者
