没有OrgDb包的非模式物种如何做功能富集?


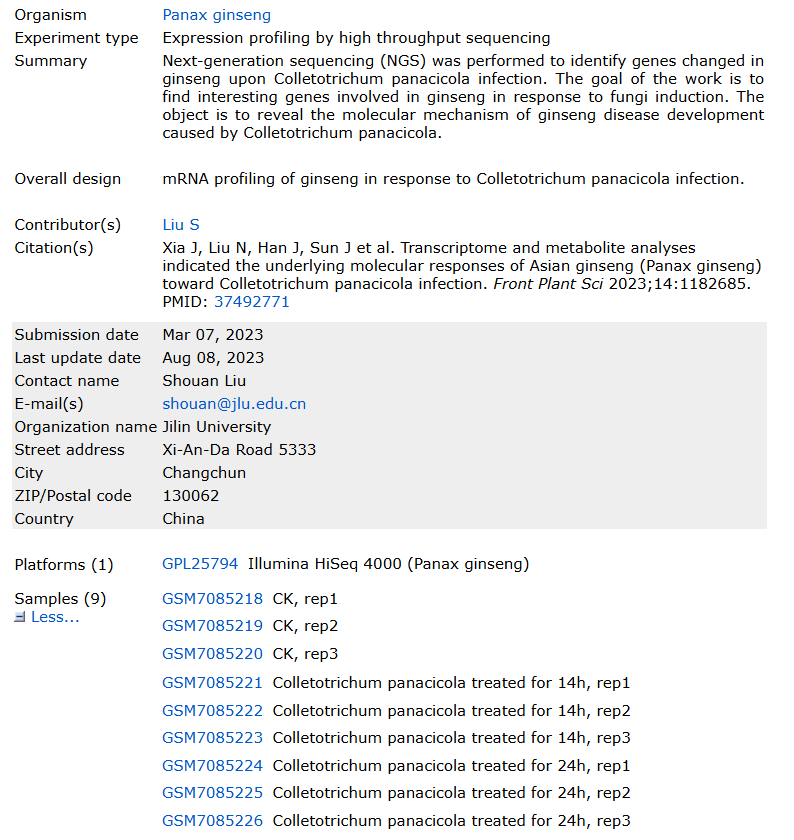
最近,我们的生信入门《转录组测序分析专题》课程进行了全面更新,里面就更新了 ,尤其是关于非模式物种的分析部分。下面来看看使用 GEO数据集 GSE226837 进行分析,物种为大家都喜爱的人参:Panax ginseng
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE226837
非模式生物(Non-model organisms)
非模式生物(Non-model organisms)是指那些在生物研究中不像模式生物那样被广泛研究和使用的生物种类。模式生物是指那些在科学研究中被广泛用作模型的生物,它们通常具有以下特点:
- 遗传信息明确:模式生物的基因组已经被完整测序,基因信息容易获取。
- 繁殖周期短:它们能够快速繁殖,便于进行遗传实验。
- 易于操作:在实验室条件下容易饲养和操作。
- 研究历史悠久:已经有大量的研究基础和资源,如基因编辑工具、研究方法等。
常见的模式生物包括:
- 果蝇(Drosophila melanogaster):在遗传学研究中非常重要。
- 小鼠(Mus musculus):在医学和生物学研究中广泛使用。
- 斑马鱼(Danio rerio):在发育生物学和毒理学研究中常用。
- 拟南芥(Arabidopsis thaliana):在植物生物学研究中作为模式植物。
- 酵母(Saccharomyces cerevisiae):在分子生物学和遗传学研究中使用。
人参(Panax ginseng)是一种非模式生物,因为它不满足上述模式生物的一些特点。人参的基因组信息可能不如模式生物那样容易获取,繁殖周期相对较长,且在实验室条件下可能不如模式生物那样容易饲养和操作。此外,人参的研究可能更侧重于其药用价值和特定生物学特性,而不是作为广泛研究的模型。然而,随着科学技术的发展,一些非模式生物的研究也在逐渐增加,它们的基因组信息和研究工具也在不断完善。人参作为一种重要的药用植物,其研究在中医药领域和植物学领域具有重要意义。
数据背景
人参炭疽病的表型以及C. panacicola FSL在2岁人参叶片上的感染表型(出现necrotic spots坏死斑点),9个样本,三种表型(对照组,病菌感染后14小时以及24小时),每组非常标准的三个生物学重复。
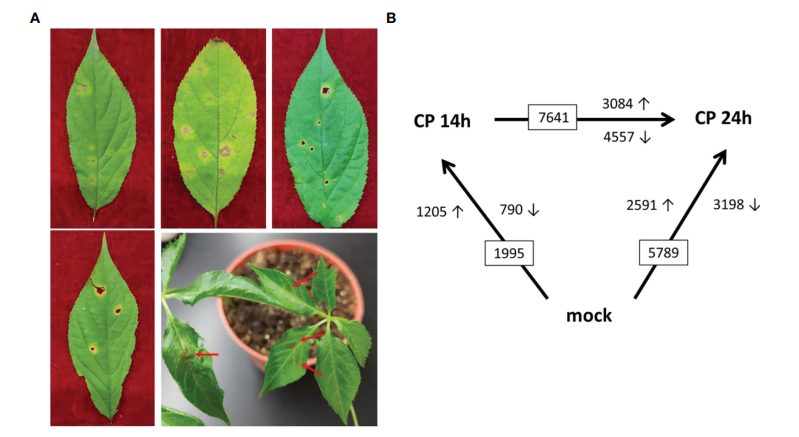
文章中做了两次 差异分析:Cp14h vs CK ; Cp24h vs CK,取差异交集:904个基因,交集温恩图,交集基因热图、交集基因KEGG功能富集
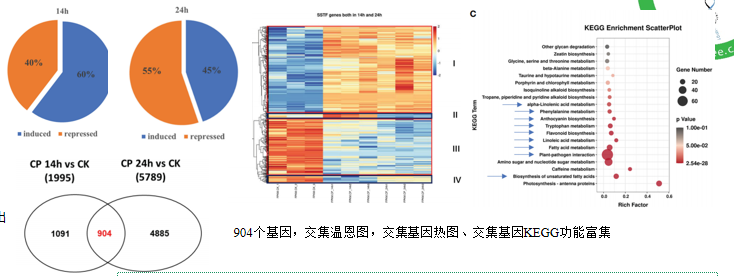
我们此次做其中一个差异分析,得到差异基因然后演示没有orgDb包如何做功能富集分析。
Cp14h vs CK 差异分析
1、获得样本分组信息:
###
### Create: Jianming Zeng
### Date: 2023-12-31
### Email: jmzeng1314@163.com
### Blog: http://www.bio-info-trainee.com/
### Forum: http://www.biotrainee.com/thread-1376-1-1.html
### CAFS/SUSTC/Eli Lilly/University of Macau
### Update Log: 2023-12-31 First version
### Update Log: 2024-11-30 by juan zhang(492482942@qq.com)
###
rm(list = ls()) # 清空当前的工作环境
options(scipen = 20) # 不以科学计数法显示
library(data.table)
library(GEOquery)
library(tidyverse)
gse_num <- "GSE226837"
dir.create(gse_num)
list.files(gse_num)
###### 1.获取样本临床信息 ###################
gset <- getGEO(gse_num, destdir = gse_num, getGPL = F)
gset[[1]]
## 根据生物学背景及研究目的人为分组
pd <- pData(gset[[1]])
head(pd)
colnames(pd)
pd$title
pd$`treatment:ch1`
##~~~分组信息编号需修改~~~
pd_select <- pd[,c("geo_accession","treatment:ch1")]
pd_select$group <- pd_select$`treatment:ch1`
pd_select$group <- gsub("Colletotrichum panacicola treated for 14h","treated_14h",pd_select$group)
pd_select$group <- gsub("Colletotrichum panacicola treated for 24h","treated_24h",pd_select$group)
table(pd_select$group)
# control treated_14h treated_24h
# 3 3 3
2、拿到表达矩阵count值和fpkm值
###### 2.获取基因表达矩阵 ###################
## read gene count and fpkm expression
data <- readxl::read_xlsx(paste0(gse_num, "/GSE226837_4_genes_fpkm_expression.xlsx"))
colnames(data)
## 提取count表达矩阵
exp <- select(data, starts_with("count")) %>%
as.data.frame()
colnames(exp) <- gsub("count.", "", colnames(exp))
rownames(exp) <- data$gene_id
head(exp)
# 过滤低表达基因:所有样本中表达都为0
keep_feature <- rowSums (exp) > 0
table(keep_feature)
exp_filter <- exp[keep_feature, ]
## 提取fpkm表达矩阵
exp_fpkm <- select(data, starts_with("FPKM")) %>%
as.data.frame()
colnames(exp_fpkm) <- gsub("FPKM.", "", colnames(exp_fpkm))
rownames(exp_fpkm) <- data$gene_id
head(exp_fpkm)
exp_fpkm_filter <- exp_fpkm[rownames(exp_filter),]
# 保存count和fpkm为rds文件
save(exp_filter, exp_fpkm_filter, pd_select, file = paste0(gse_num, '/exp.Rdata'))
# 保存fpkm为表格文件
exp_fpkm_filter <- exp_fpkm_filter %>%
rownames_to_column("ID")
write.table(exp_fpkm_filter, file = paste0(gse_num, "/exp_fpkm.xls"), row.names = F,sep = "\t",quote = F)
# 保存count为表格文件
exp_filter <- exp_filter %>%
rownames_to_column("ID")
write.table(exp_filter, file = paste0(gse_num, "/exp_counts.xls"), row.names = F,sep = "\t",quote = F)
3、获取所有基因id与KEGG通路以及GO通路对应关系
我们能够做功能富集主要是作者提供的数据比较特殊,表格 GSE226837_4_genes_fpkm_expression.xlsx 中提供了所有基因ID的KEGG通路与GO数据库Term的注释:
如果你是拿到的公司给你的转录组标准分析报告,那肯定是有这个表格结果的。
如果没有的话,想要拿到每个基因的不同数据库的功能注释结果,就需要做不同数据库的blast基因序列比对来对基因进行注释,这个部分我们后面介绍。
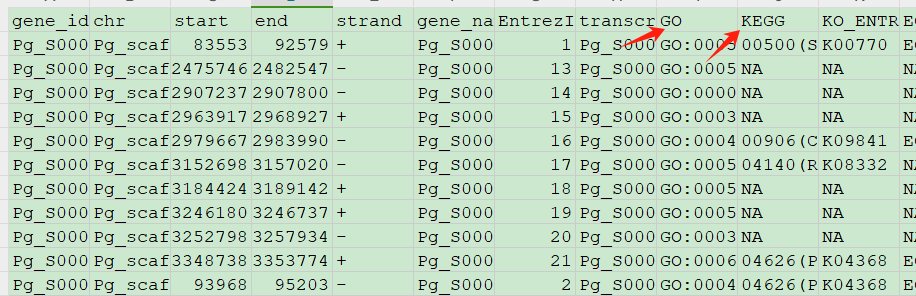
处理这个表格拿到 基因id与KEGG通路以及GO通路对应关系:
## GO 数据库
go2gene <- list()
# KEGG 数据库
kegg2gene <- list()
for(i in 1:nrow(data)) {
#i <- 1
geneid <- data$gene_id[i]
# go term
temp <- unlist(str_split(data$GO[i],pattern = ";"))
temp <- str_split(temp,pattern = "\\(", simplify = T, n = 2)
goid <- temp[,1]
go_term <- gsub("\\)","",temp[,2])
res <- data.frame(geneid, goid, go_term)
go2gene[[i]] <- res
# kegg pathway
temp <- unlist(str_split(data$KEGG[i],pattern = ";"))
temp <- str_split(temp,pattern = "\\(", simplify = T, n = 2)
keggid <- temp[,1]
kegg_path <- gsub("\\)","",temp[,2])
res <- data.frame(geneid, keggid, kegg_path)
kegg2gene[[i]] <- res
print(i)
}
go2gene_filnal <- do.call(rbind, go2gene)
go2gene_filnal <- go2gene_filnal[go2gene_filnal$goid!="NA",]
go2gene_filnal <- go2gene_filnal[order(go2gene_filnal$goid),]
kegg2gene_filnal <- do.call(rbind, kegg2gene)
kegg2gene_filnal <- kegg2gene_filnal[kegg2gene_filnal$keggid!="NA", ]
kegg2gene_filnal <- kegg2gene_filnal[order(kegg2gene_filnal$keggid),]
save(go2gene_filnal, kegg2gene_filnal, file = paste0(gse_num, '/path2gene.Rdata'))
write.table(go2gene_filnal, file = paste0(gse_num, "/go2gene.xls"), row.names = F,sep = "\t",quote = F)
write.table(kegg2gene_filnal, file = paste0(gse_num, "/kegg2gene.xls"), row.names = F,sep = "\t",quote = F)
基因id与KEGG通路:
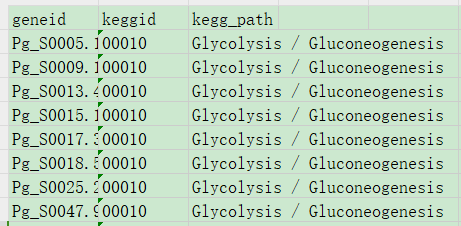
GO通路对应关系:
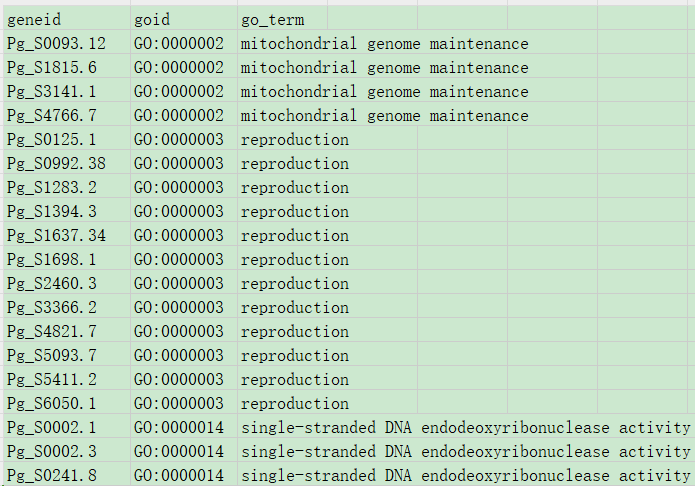
如果有OrgDb包,Bioconductor项目的一部分
它提供了一种方便的方式来存储和检索特定物种的注释信息。这些包通常包含了基因、转录本、蛋白质的详细信息,以及它们与数据库如Entrez、Ensembl、UniProt等的对应关系,还有基因本体(GO)注释等。OrgDb包不仅限于模式生物,也包括了一些经济动物和作物。比如:
- 模式生物:人(org.Hs.eg.db)、小鼠(org.Mm.eg.db)、拟南芥(org.At.tair.db)、果蝇(org.Dm.eg.db)、斑马鱼(org.Dr.eg.db)等。
- 经济动物:牛(org.Bt.eg.db)、犬(org.Cf.eg.db)、猪(org.Ss.eg.db)等。
- 经济作物:需要自行在Bioconductor的OrgDb包列表中查找特定作物的OrgDb包。
如果你需要为特定的非模式生物寻找或构建OrgDb包,可以使用AnnotationHub来搜索现有的包,或者使用AnnotationForge来构建新的OrgDb包。此外,有些非模式生物可能在AnnotationHub中存在相应的注释信息,可以通过这个平台获取。
4、差异分析
rm(list = ls()) ## 魔幻操作,一键清空~
library(limma)
library(edgeR)
library(DESeq2)
getOption('timeout')
options(timeout=10000)
## 加载数据
load('./GSE226837/exp.Rdata')
symbol_matrix <- exp_filter
symbol_matrix[1:4,1:4]
dim(symbol_matrix)
# 生成一个group_list
group_list <- colnames(exp_fpkm_filter)
group_list <- substr(group_list,1, nchar(group_list)-1)
group_list
table(group_list)
vs <- 'result/CP_14h-vs-CK'
vs
dir.create(vs)
# 提取对应样本的矩阵和分组
kp <- group_list %in% c('CK','CP_14h')
table(kp)
symbol_matrix <- symbol_matrix[,kp]
group_list <- group_list[kp]
group_list
table(group_list)
group_list <- ifelse(group_list=='CK','control','case')
group_list <- factor(group_list,levels = c('control','case' ))
group_list
save(symbol_matrix,group_list,file = paste0(vs,'/symbol_matrix.Rdata'))
###################################################
## Deseq2
# 差异分析count矩阵
exprSet <- symbol_matrix
class(exprSet)
# 差异分组矩阵
colData <- data.frame(row.names=colnames(exprSet), group_list=group_list)
colData
levels(group_list)[2]
levels(group_list)[1]
# deseq2差异分析
dds <- DESeqDataSetFromMatrix(countData = exprSet, colData = colData, design = ~ group_list)
dds <- DESeq(dds)
res <- results(dds, contrast=c("group_list",levels(group_list)[2], levels(group_list)[1]) )
# 对差异结果按照校正后的pvalue排序
resOrdered <- res[order(res$padj),]
head(resOrdered)
DEG <- as.data.frame(resOrdered)
# na.omit函数去掉可能存在na值的行
DEG_deseq2 <- na.omit(DEG)
# 添加调控信息
DEG_deseq2$regulated <- "Normal"
DEG_deseq2$regulated[DEG_deseq2$log2FoldChange>1 & DEG_deseq2$padj<0.05] <- "Up"
DEG_deseq2$regulated[DEG_deseq2$log2FoldChange< -1 & DEG_deseq2$padj<0.05] <- "Down"
table(DEG_deseq2$regulated)
# 保存结果
save(DEG_deseq2, file = paste0(vs, '/DEG_deseq2.Rdata' ))
GO数据库与KEGG数据库功能富集
1、KEGG数据库 通路富集
使用 enricher 函数做功能富集分析,TERM2GENE 参数指定前面处理好的通路与基因关系
rm(list = ls())
library(clusterProfiler)
library(ggplot2)
library(stringr)
# 以 Deseq2 差异结果为例
load( file = 'result/CP_14h-vs-CK/DEG_deseq2.Rdata' )
colnames(DEG_deseq2)
# 得到上调与下调基因
gene_up <- rownames( DEG_deseq2[DEG_deseq2$regulated=="Up",] )
gene_down <- rownames( DEG_deseq2[DEG_deseq2$regulated=="Down",] )
length(gene_up);length(gene_down)
head(gene_up);head(gene_down)
write.table(gene_up,file = 'result/CP_14h-vs-CK/gene_up.txt',quote = F,col.names = F,row.names = F)
write.table(gene_down,file = 'result/CP_14h-vs-CK/gene_down.txt',quote = F,col.names = F,row.names = F)
## 2.3.1 KEGG----
# 读取之前处理的 KEGG通路与基因id的关系
kegg_infor <- read.table(file = "GSE226837/kegg2gene.xls", header = T,sep = "\t")
head(kegg_infor)
kegg_term <- kegg_infor[,c(3,1)]
head(kegg_term) #第一列为通路,第二列为基因
# 上调基因富集,使用enricher函数做功能富集分析,TERM2GENE参数制定通路与基因关系
kk.up <- enricher(gene = gene_up,pvalueCutoff = 0.9, qvalueCutoff =0.9, TERM2GENE=kegg_term)
head(kk.up)[,1:6]
dotplot(kk.up)
barplot(kk.up)
# 下调基因富集,使用enricher函数做功能富集分析,TERM2GENE参数制定通路与基因关系
kk.down <- enricher(gene = gene_down,pvalueCutoff = 0.9, qvalueCutoff =0.9, TERM2GENE=kegg_term)
head(kk.down)[,1:6]
dotplot(kk.down)
barplot(kk.down)
## 个性化绘图
kegg_down_dt <- as.data.frame(kk.down)
kegg_up_dt <- as.data.frame(kk.up)
down_kegg <- kegg_down_dt[kegg_down_dt$pvalue<0.01,];down_kegg$group=-1
up_kegg <- kegg_up_dt[kegg_up_dt$pvalue<0.01,];up_kegg$group=1
# 定义一个绘图函数
kegg_plot <- function(up_kegg,down_kegg){
dat=rbind(up_kegg,down_kegg)
colnames(dat)
dat$pvalue = -log10(dat$pvalue)
dat$pvalue=dat$pvalue*dat$group
dat=dat[order(dat$pvalue,decreasing = F),]
g_kegg <- ggplot(dat, aes(x=reorder(Description,order(pvalue, decreasing = F)), y=pvalue, fill=group)) +
geom_bar(stat="identity") +
scale_fill_gradient(low="#34bfb5",high="#ff6633",guide = 'none') +
scale_x_discrete(name ="") +
scale_y_continuous(name ="log10P-value") +
ggtitle("KEGG Enrichment") +
coord_flip() + theme_bw()+
theme(plot.title = element_text(size = 15,face = 'bold',hjust = 0.5),
axis.text = element_text(size = 12,face = 'bold'),
panel.grid = element_blank())
}
g_kegg <- kegg_plot(up_kegg,down_kegg)
print(g_kegg)
ggsave('result/CP_14h-vs-CK/kegg_up_down.pdf',height = 7,width = 9)
结果如下:
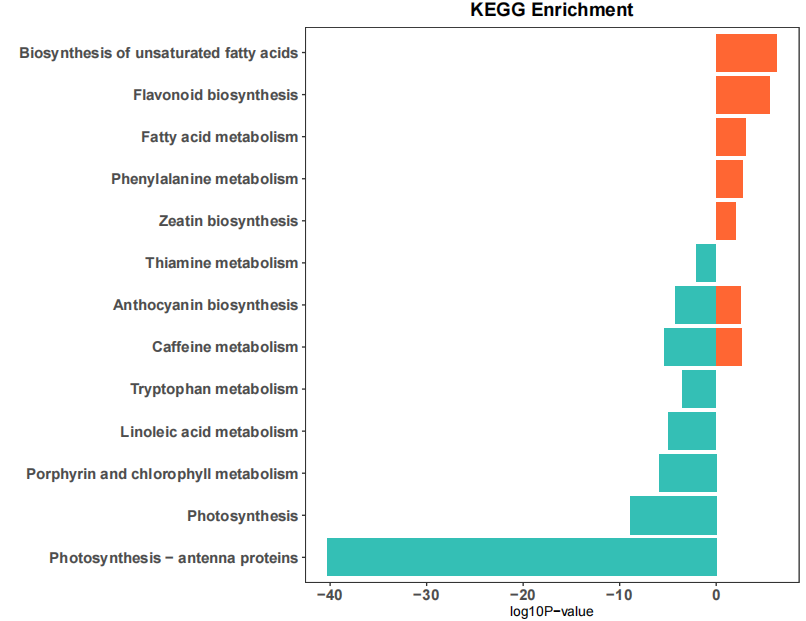
2、GO数据库 通路富集
同样也是 使用 enricher 函数做功能富集分析,TERM2GENE 参数指定前面处理好的通路与基因关系
## 2.3.2 GO----
# 读取之前处理的 GO通路与基因id的关系
go_infor <- read.table(file = "GSE226837/go2gene.xls", header = T,sep = "\t", quote = "\"")
head(go_infor)
go_term <- go_infor[,c(3,1)]
head(go_infor) #第一列为通路,第二列为基因
# 上调基因富集,使用enricher函数做功能富集分析,TERM2GENE参数制定通路与基因关系
go.up <- enricher(gene = gene_up,pvalueCutoff = 0.9, qvalueCutoff =0.9, TERM2GENE=go_term)
head(go.up)[,1:6]
# label_format参数解决绘图中标签重叠问题
dotplot(go.up, label_format=100)
barplot(go.up, label_format = 100)
# 下调基因富集,使用enricher函数做功能富集分析,TERM2GENE参数制定通路与基因关系
go.down <- enricher(gene = gene_down,pvalueCutoff = 0.9, qvalueCutoff =0.9, TERM2GENE=go_term)
head(go.down)[,1:6]
# label_format参数解决绘图中标签重叠问题
dotplot(go.down, label_format=100)
ggsave(filename = 'result/CP_14h-vs-CK/go_up.pdf',height = 7,width = 9)
barplot(go.down, label_format = 100)
ggsave(filename = 'result/CP_14h-vs-CK/go_down.pdf',height = 7,width = 9)
结果如下 :
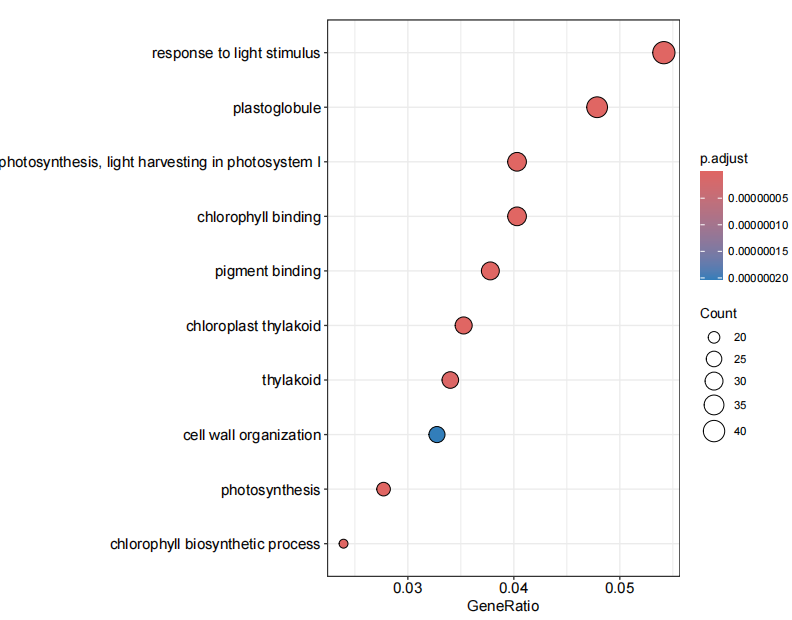
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-01-07,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号