全网最佳WGCNA分析教程 | 更新:增加可视化及任意基因共表达网络Cytoscape作图

全网最佳WGCNA分析教程 | 更新:增加可视化及任意基因共表达网络Cytoscape作图

简说基因
发布于 2025-12-24 14:21:47
发布于 2025-12-24 14:21:47
本教程带你零代码进行 WGCNA 分析。无需配置任何环境,上传数据即可一键分析。也可以分步骤完成,做到知其然,并知其所以然。最后,我们可以输出任意基因的共表达网络,用于 Cytoscape 作图。
WGCNA(Weighted Gene Co-expression Network Analysis,加权基因共表达网络分析)是一种基于基因表达数据(如 RNA-seq、微阵列)挖掘基因间共表达模式,并关联到表型特征的有用方法。
WGCNA 数据分析大致遵循下图所示的步骤,本文会涉及此图的大部分内容。我们先从一键完成开始,然后再介绍分步骤进行 WGCNA 分析的方法。
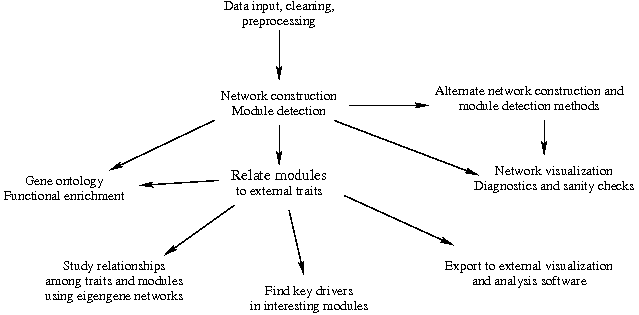
一键完成 WGCNA 分析
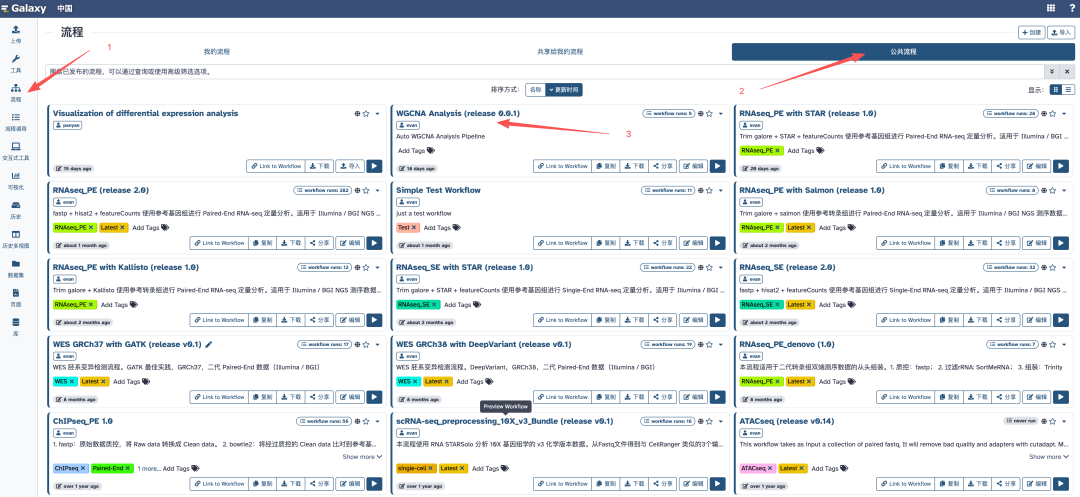
我们首先进入网站:https://usegalaxy.cn,依次点击:流程 > 公共流程 > WGCNA Analysis (release 0.0.1),如图:
点击流程名称:WGCNA Analysis (release 0.0.1),可以预览一下流程所要执行的步骤:
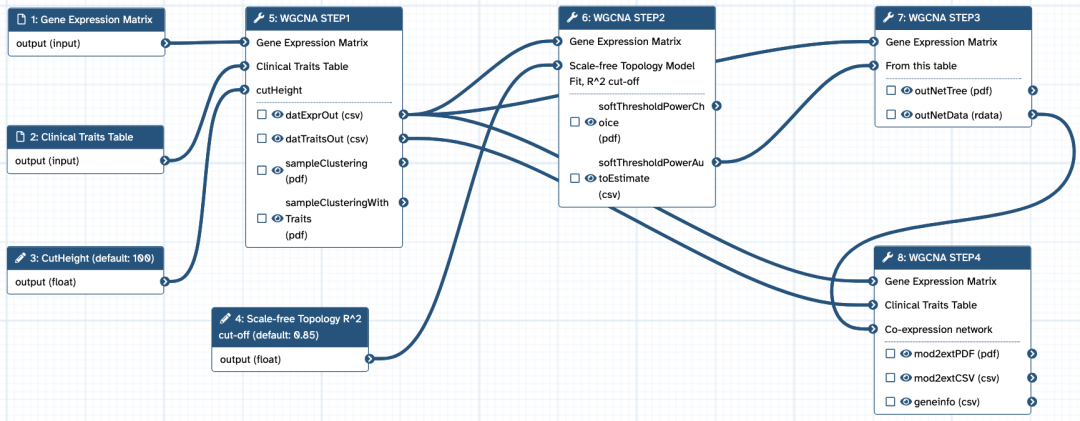
可以看到,该流程主要由 4 个步骤组成:STEP1、STEP2、STEP3、STEP4。
点击“运行流程”按钮,进入这样一个界面:
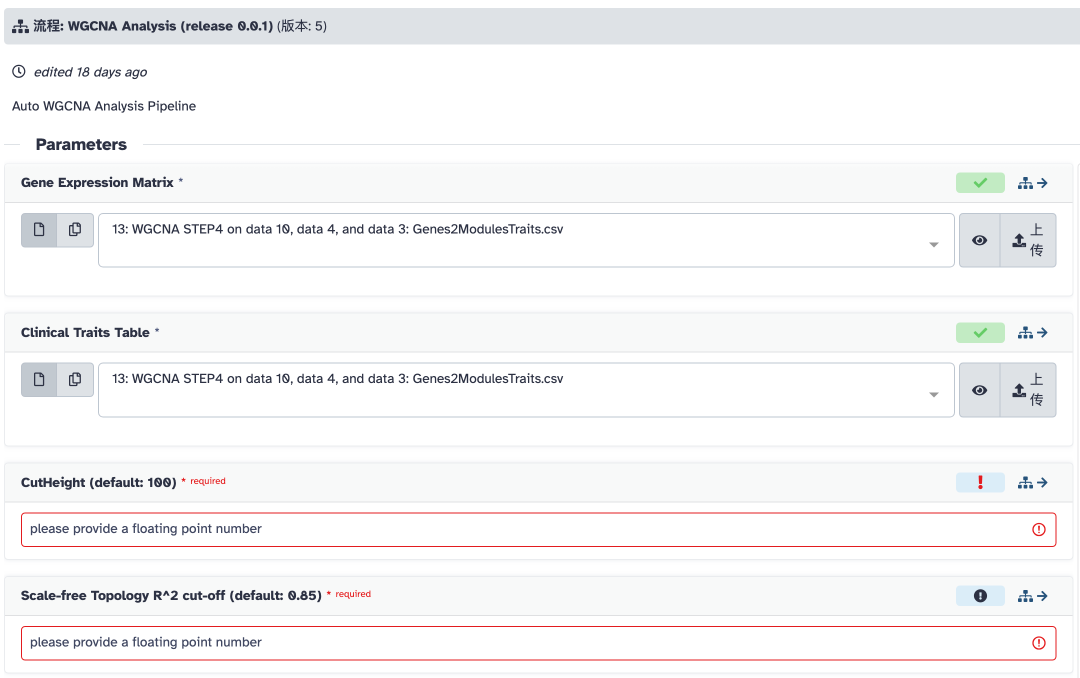
流程要求输入 2 个文件和 2 个数值:
- • Gene Expression Matrix:基因表达量矩阵(行是样本,列是基因。第 1 列是样本编号,其他列是基因的表达量)。要求是 csv 文件,即英文逗号分隔的文件。
- • Clinical Traits Table:特征表格文件(行是样本,列是特征。第 1 列是样本编号,其他列是样本的各种特征,如身高、体重等)。也要求是 csv 文件。
- • CutHeight:去除异常样本的截断值,默认是 100,不去除异常样本。
- • Scale-free Topology R^2 cut-off:无标度拓扑结构 R 方截断值,通常应不低于 0.85。
参数设置
我们用两个测试文件进行演示。测试文件位置:
左侧激活栏 > 库 > RNA-seq > WGCNA:
- • datExpr0.csv,表达量矩阵
- • datTraits0.csv,样本特征表
我们将两个文件添加到历史记录中,然后设置流程参数,如图:
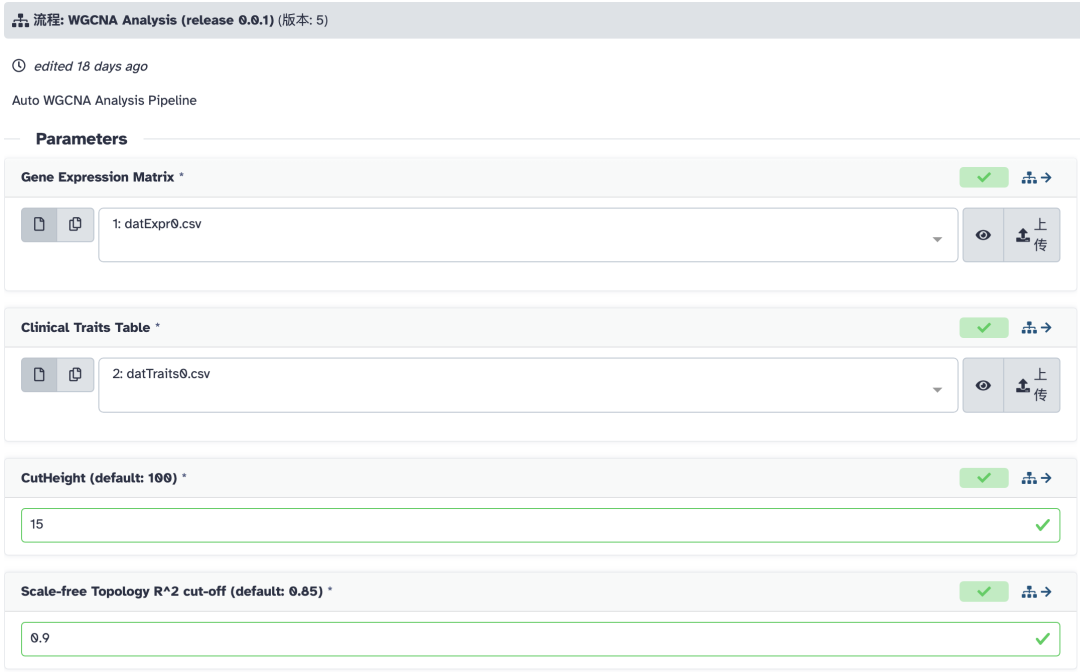
注意表达量矩阵和样本特征表格的位置不要弄错了。另外我们还设置了两个参数:
- • CutHeight:15
- • Scale-free Topology R^2 cut-off:0.9
因为是测试数据,我们知道这两个参数的合理值。如果你是分析自己的数据,可以先将 CutHeight 设置为:100,R^2 值设置为:0.85。
运行流程
点“运行流程”,等待结果就可以了。
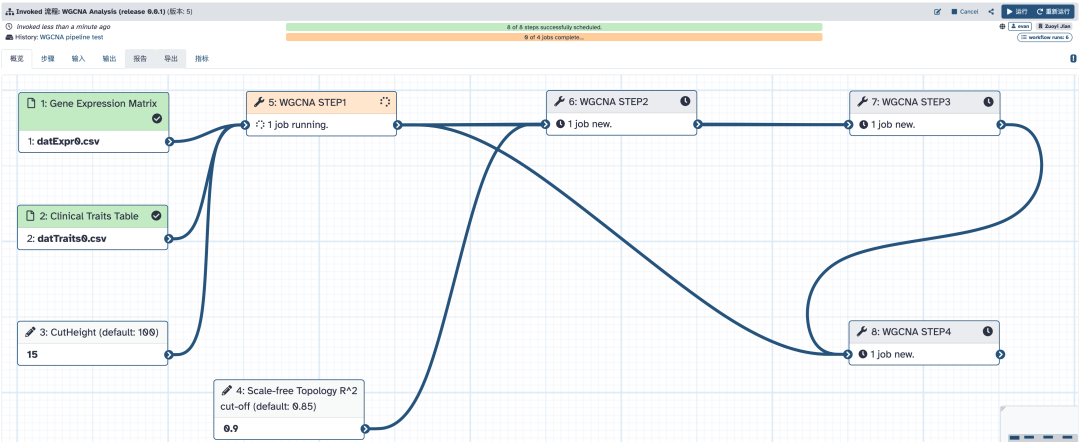
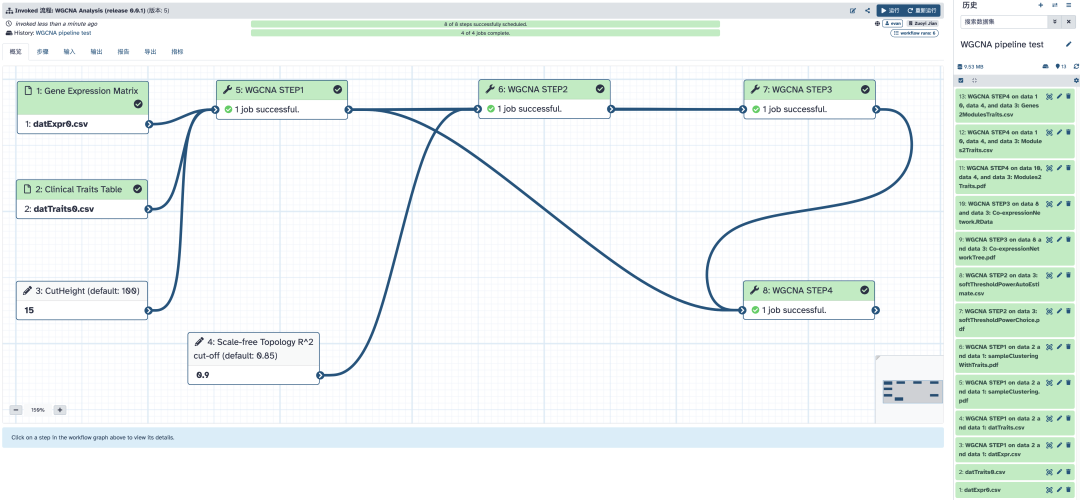
查看结果
流程运行完毕,所有结果文件都在右侧的历史中,具体的文件内容我们在这里不展开讨论,后面分步执行 WGCNA 的时候再细说。
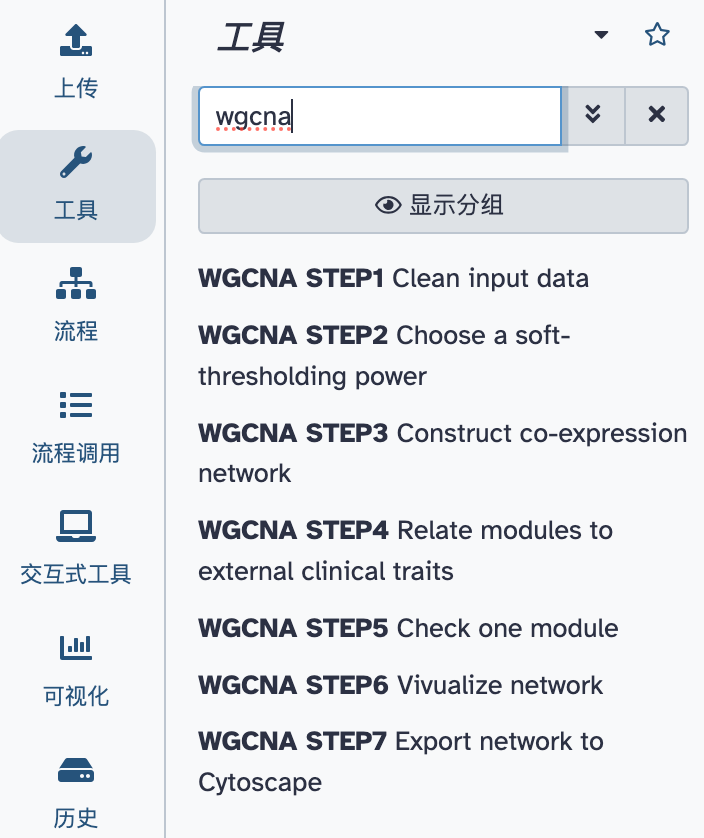
分步完成 WGCNA 分析
在平台“工具”的搜索框中,我们键入:wgcna,下方即会出现 WGCNA STEP 开头的 7 个工具。我们现在来分别执行这几个工具,从而帮助大家理解 WGCNA 的分析过程。
WGCNA STEP1
第一步目的是弄清楚有哪些异常样本,从而去除掉,防止影响后续分析。
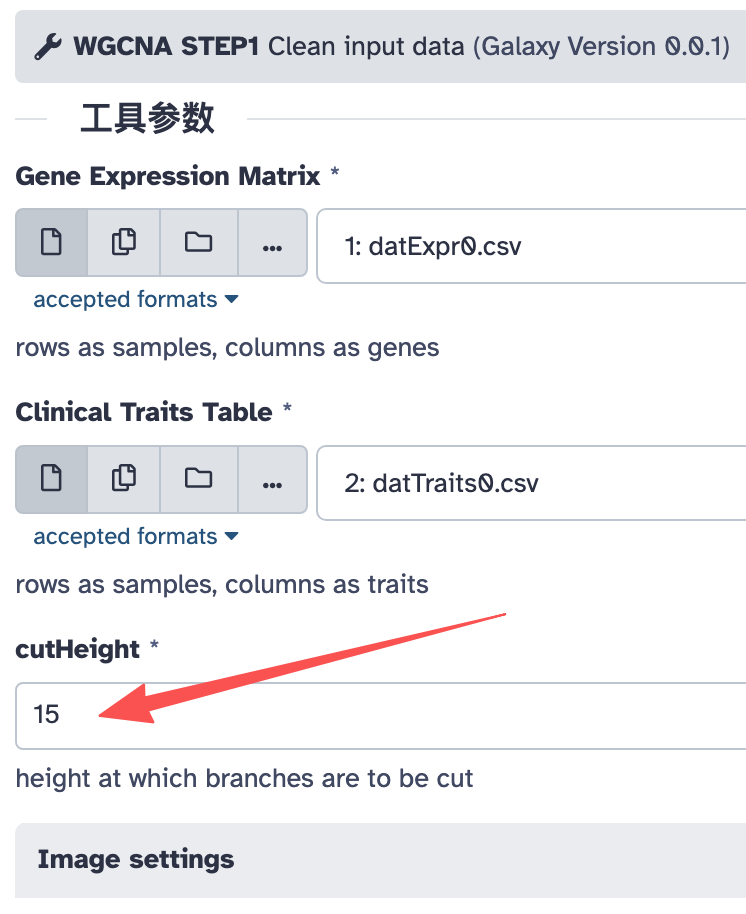
我们先简单看一下。按下图设置参数,运行:

第一步执行完成后,我们看到右侧多了 4 个文件:

- • datExpr.csv,是清除异常样本后的表达量矩阵,后续分析步骤使用该矩阵
- • datTraits.csv,是清除异常样本后的样本特征表,后续分析步骤使用该表
- • sampleClustering.pdf,样本聚类结果,以检测异常样本
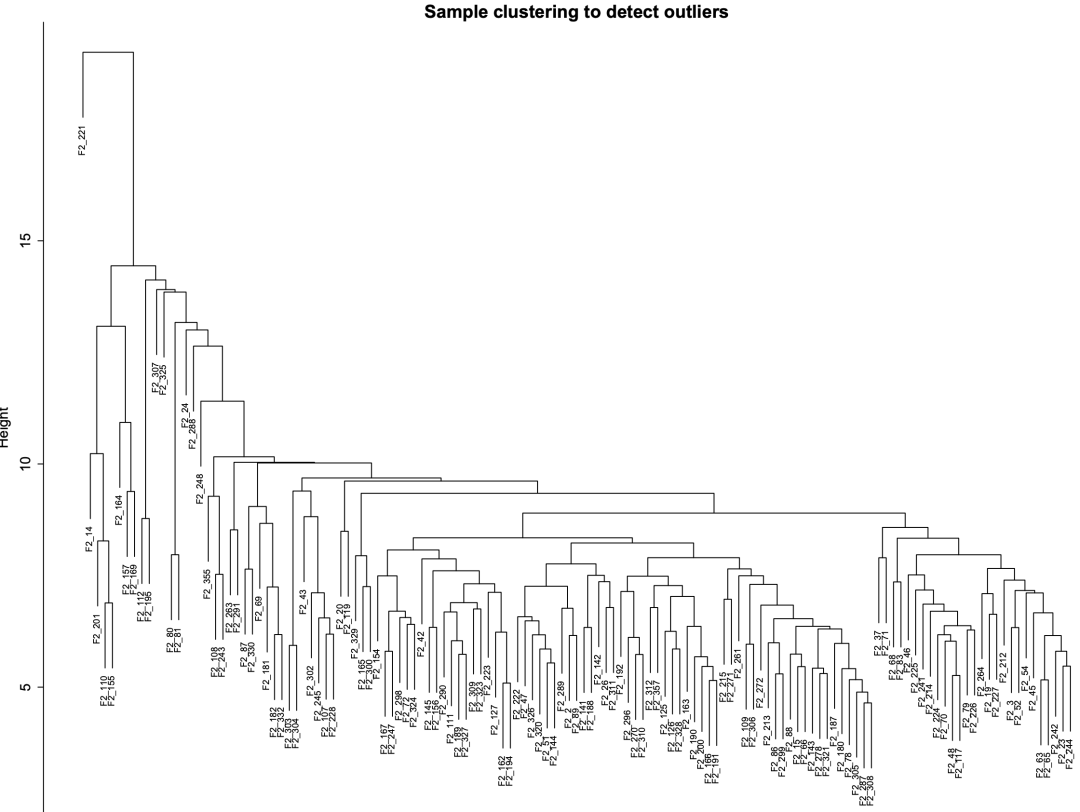
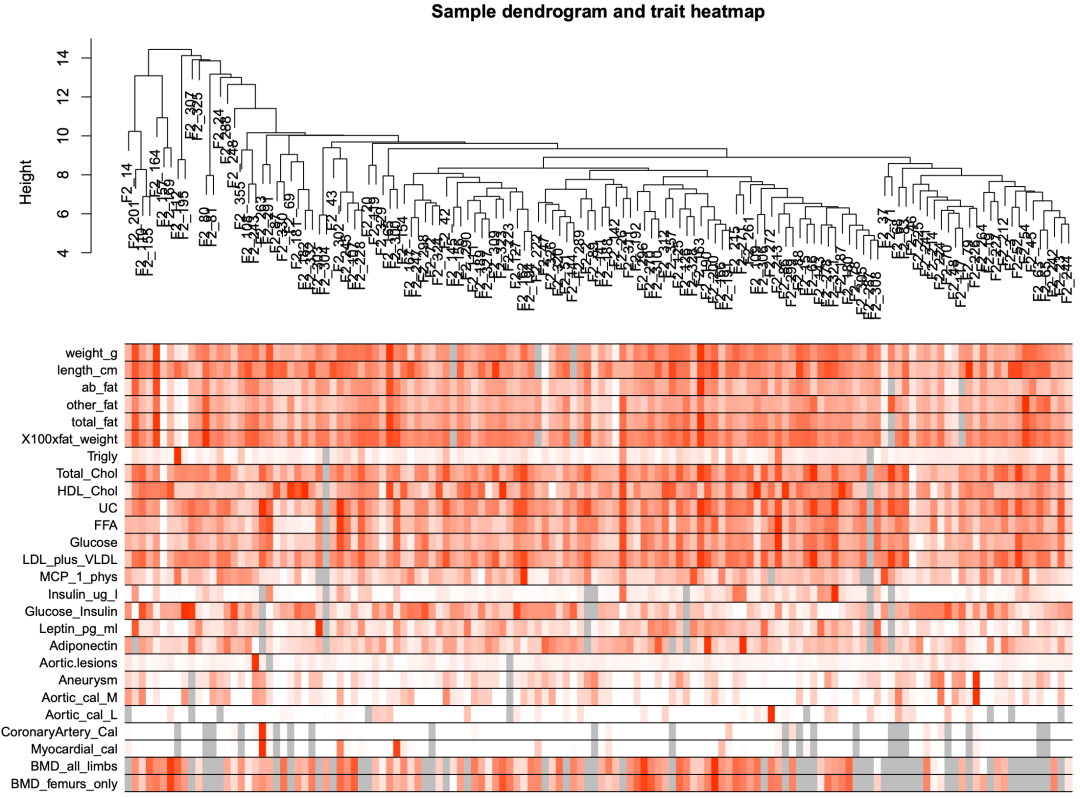
- • sampleClusteringWithTraits.pdf,样本聚类,并关联临床信息
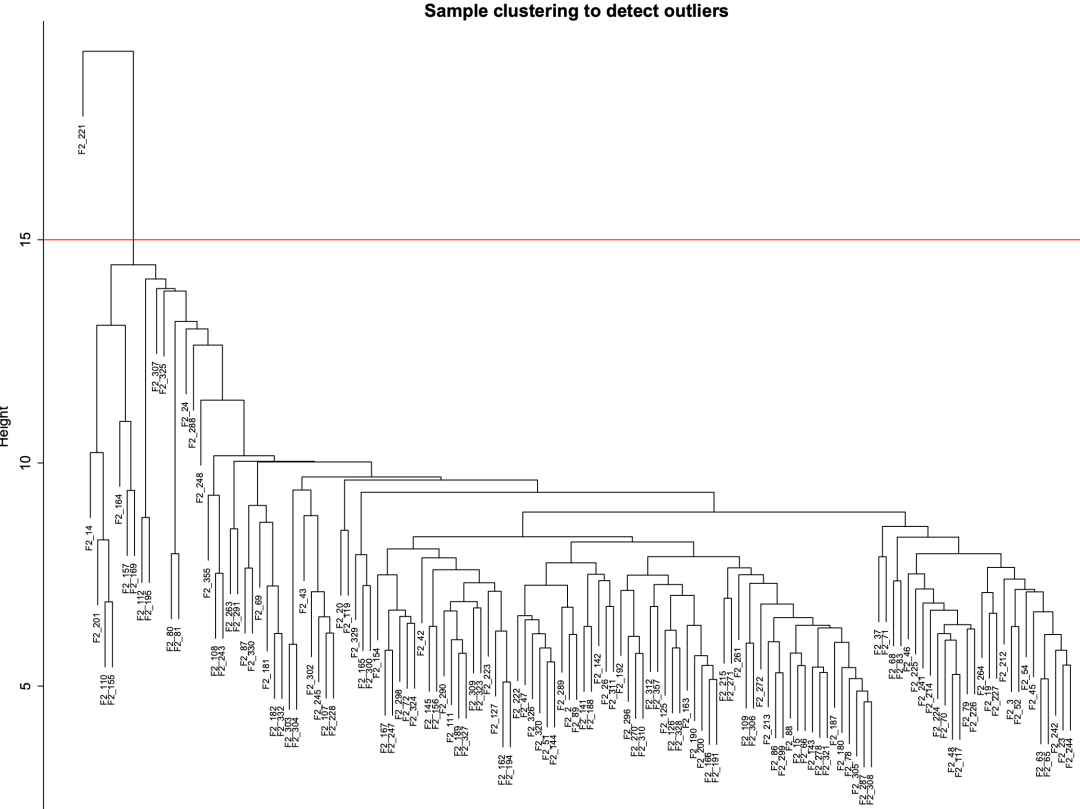
通过 sampleClustering.pdf 图片可以看到,有一个离群样本:F2_221,它在纵坐标 15 的上方,我们可以将 cutHeight 值设为 15,过滤掉该样本。
重新设置 cutHeight 后的 sampleClustering.pdf 图:
WGCNA STEP2
这一步是选择软阈值。我们设置 R^2 值为:0.9,即认为 R^2 达到 0.9 的基因才认为它们是相连的。
注意这里使用的表达量矩阵是我们最后过滤好的:历史中第 7 号文件,你可以为它改个容易识别的名字,我这里就不改了。
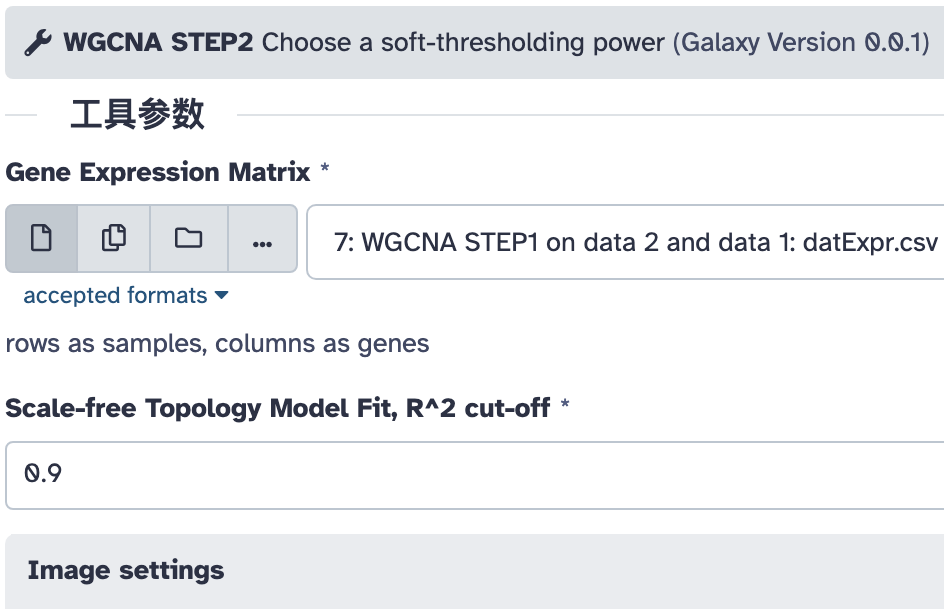
输出 2 个文件:
- • softThresholdPowerChoice.pdf,软阈值选择参考图片
- • softThresholdPowerAutoEstimate.csv,WGCNA 自动选择的软阈值
查看软阈值选择图片:
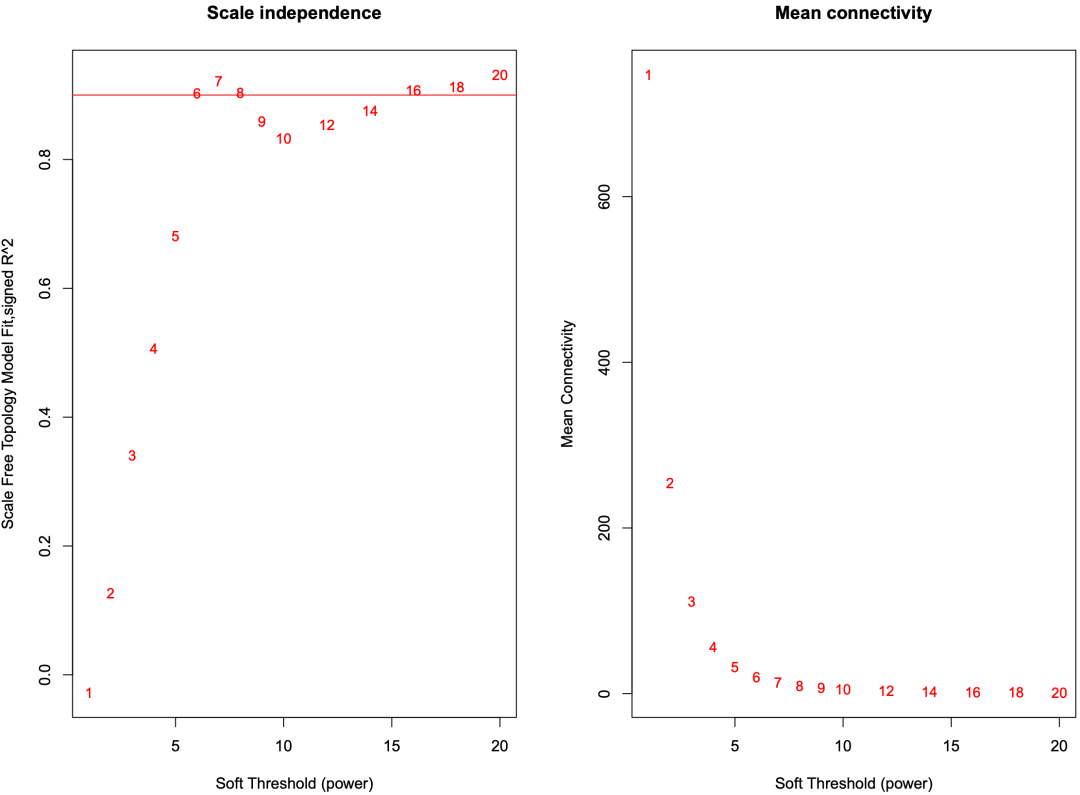
左图,随着软阈值逐渐增大,R^2 也逐渐变大。但当软阈值大于 6 时,R^2 几乎停止增加,甚至还有下降,但逐渐趋于平稳。
右图,随着软阈值的逐渐增大,基因之间的平均连通性逐渐变小。但当软阈值大于 6 时,连通性下降趋于平稳。
结合左右图,我们可以认为软阈值为 6 是比较合适的,这跟 WGCNA 自动判断得到的值一样。
WGCNA STEP3
这一步构建基因的加权共表达网络。
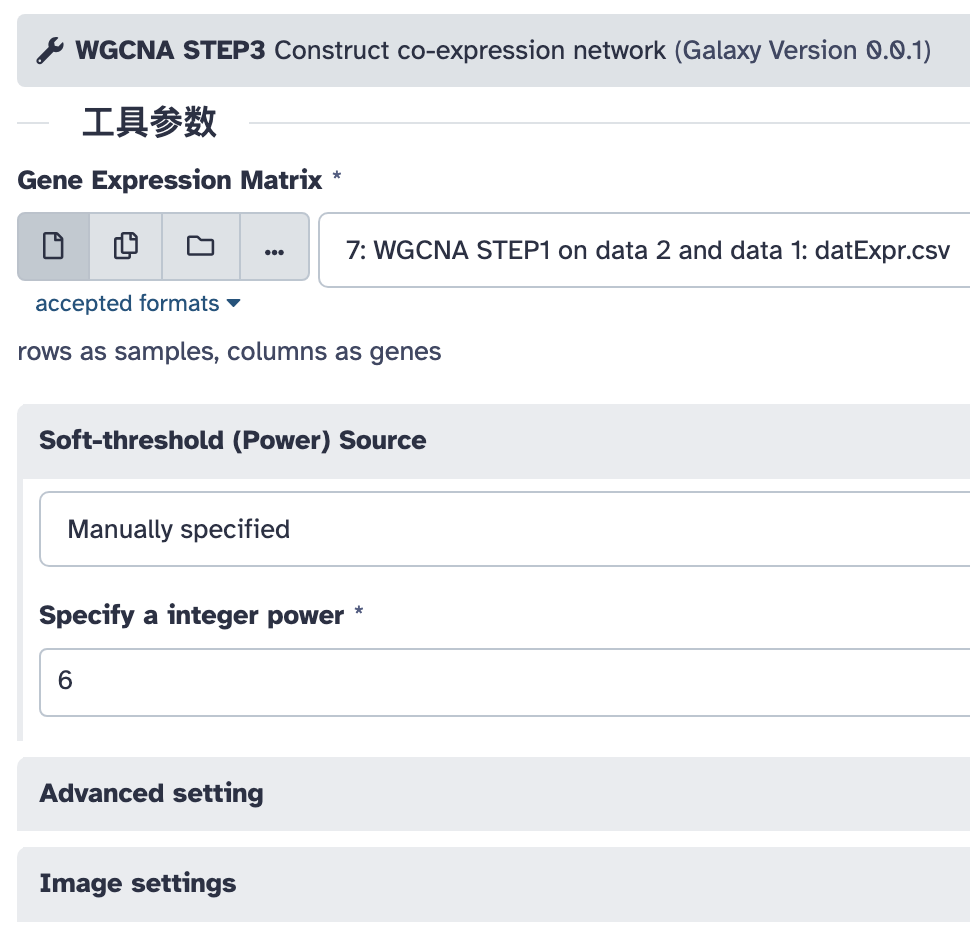
这里软阈值可以使用上一步输出的软阈值结果文件,也可以人为指定一个,我们指定为:6。
运行完成后,激动人心的时刻到了。我们得到了基因模块的层次聚类图:Co-expressionNetworkTree.pdf,这是可以放到文章当中的:
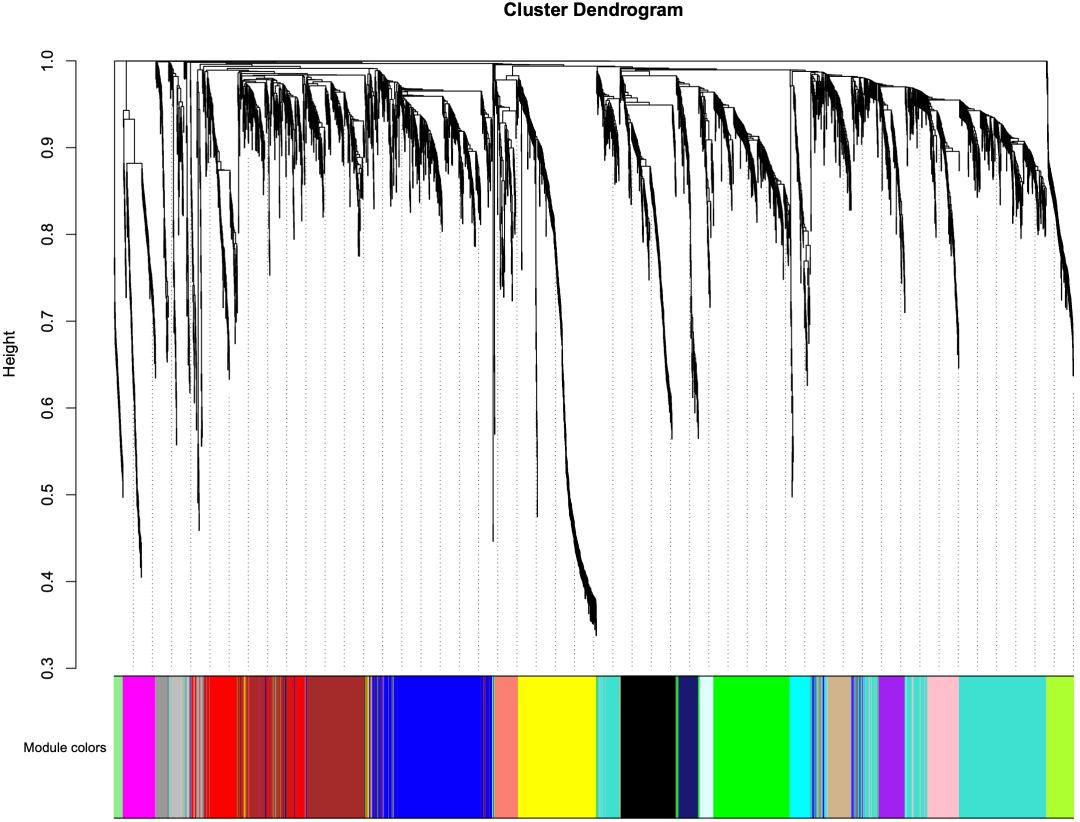
另外一个结果文件是:Co-expressionNetwork.RData,共表达网络 R 语言对象,后续要用。
WGCNA STEP4
这一步是将基因模块关联到样本的特征。注意这里的基因表达矩阵和样本特征表要选择前面第一步过滤好的,本教程中是历史中的第 7 号、8 号两个文件。另外还要输入上一步构建好的共表达网络。
又得到一个发表级图:Modules2Traits.pdf
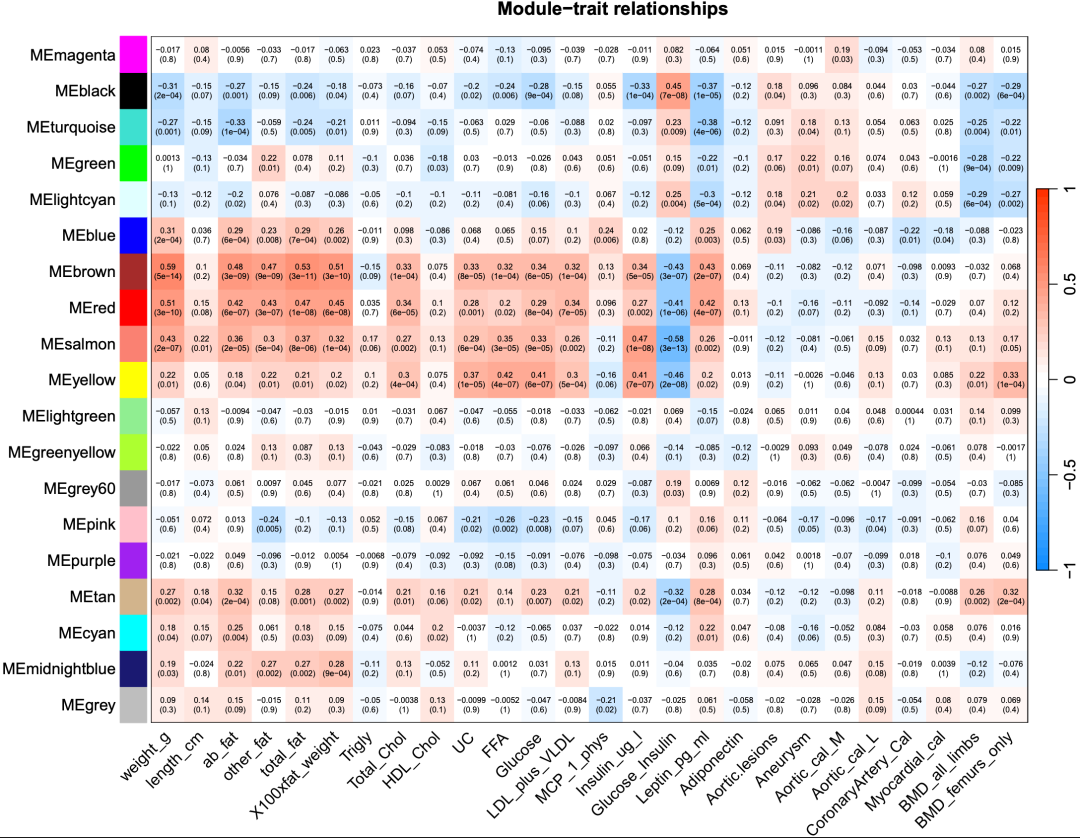
这个图展示了基因模块与临床特征的关联:
- • 模块的颜色代表关联程度的大小,越偏红,关联程度越大
- • 括号中是 p 值,越小,关联结果越可靠
我们可以基于此图筛选感兴趣的模块。
由于上图是综合了模块与临床特征的相关性,以及基因与临床特征的相关性,因此我们也输出了两个相关性表,供后续使用:
- • Modules2Traits.csv
- • Genes2ModulesTraits.csv
WGCNA STEP5
这一步是选择具体某一个模块。比如我们通过模块与特征的热图可以看出:brown 模块与 weight_g 相关性达到了 0.59,我们如果对这一个模块感兴趣,可以单独查看该模块的情况。
参数设置:
- • 输入表格:Genes2ModulesTraits.csv,即上一步得到的记录了基因与模块以及基因与特征的相关性表格
- • 模块颜色:brown
- • Column Module Membership:选择基因与模块相关性所在的列,c9
- • Column Gene Significance:选择基因与特征相关性所在的列,c41
结果:
- • gene2moduleTraitsScatter.pdf,散点图。X 轴:基因与模块的相关性;Y 轴:基因与特征的相关性
- • gene2moduleTraitsScatter.csv,用于绘制散点图的表格。还列出了模块内的所有基因
查看散点图:
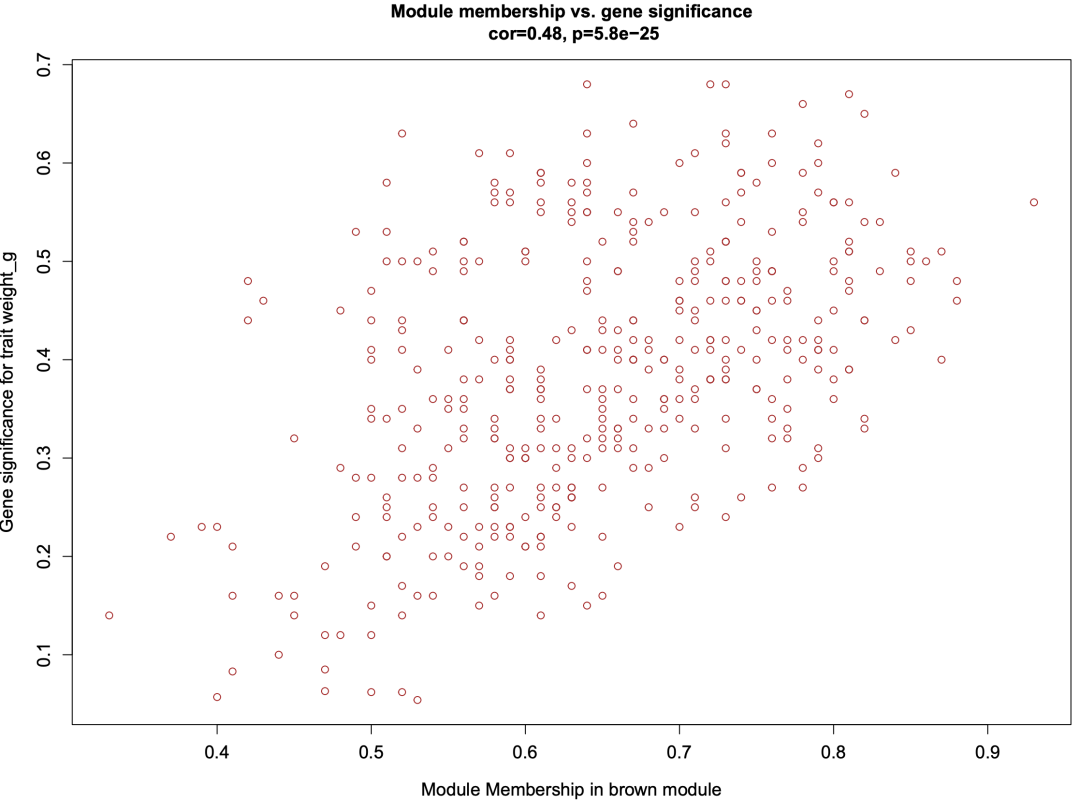
可以看到,基因与模块的相关性越强,则基因与特征之间的相关性也越强。后续可以筛选图中右上角的基因进一步分析,比如可以筛选基因与模块的相关性大于 0.8,以及基因与特征的相关性大于 0.6 的基因,用于 GO、KEGG、GSEA 等富集分析。
WGCNA STEP6
这一步我们再为 WGCNA 增加一些可视化功能。
参数设置:
- • Gene Expression Matrix:过滤好的基因表达量矩阵
- • Sample Traits Table:过滤好的样本特征表格
- • Select column for visualization:选择所关心的特征所在的列
- • Co-expression network:前面构建的基因共表达网络
- • Soft-threshold (Power) Source:软阈值,可以手动填入,也可以用前面自动计算的
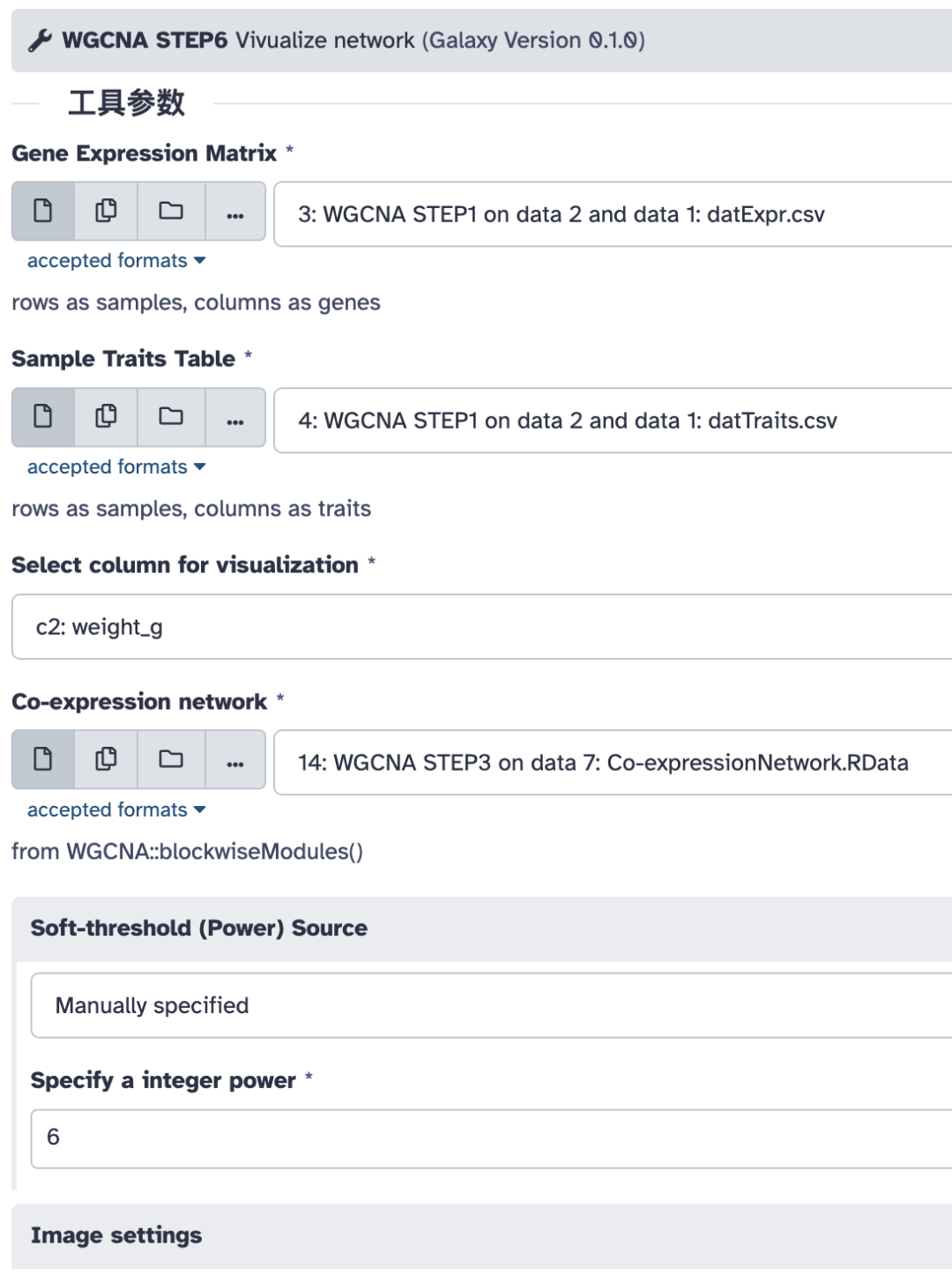
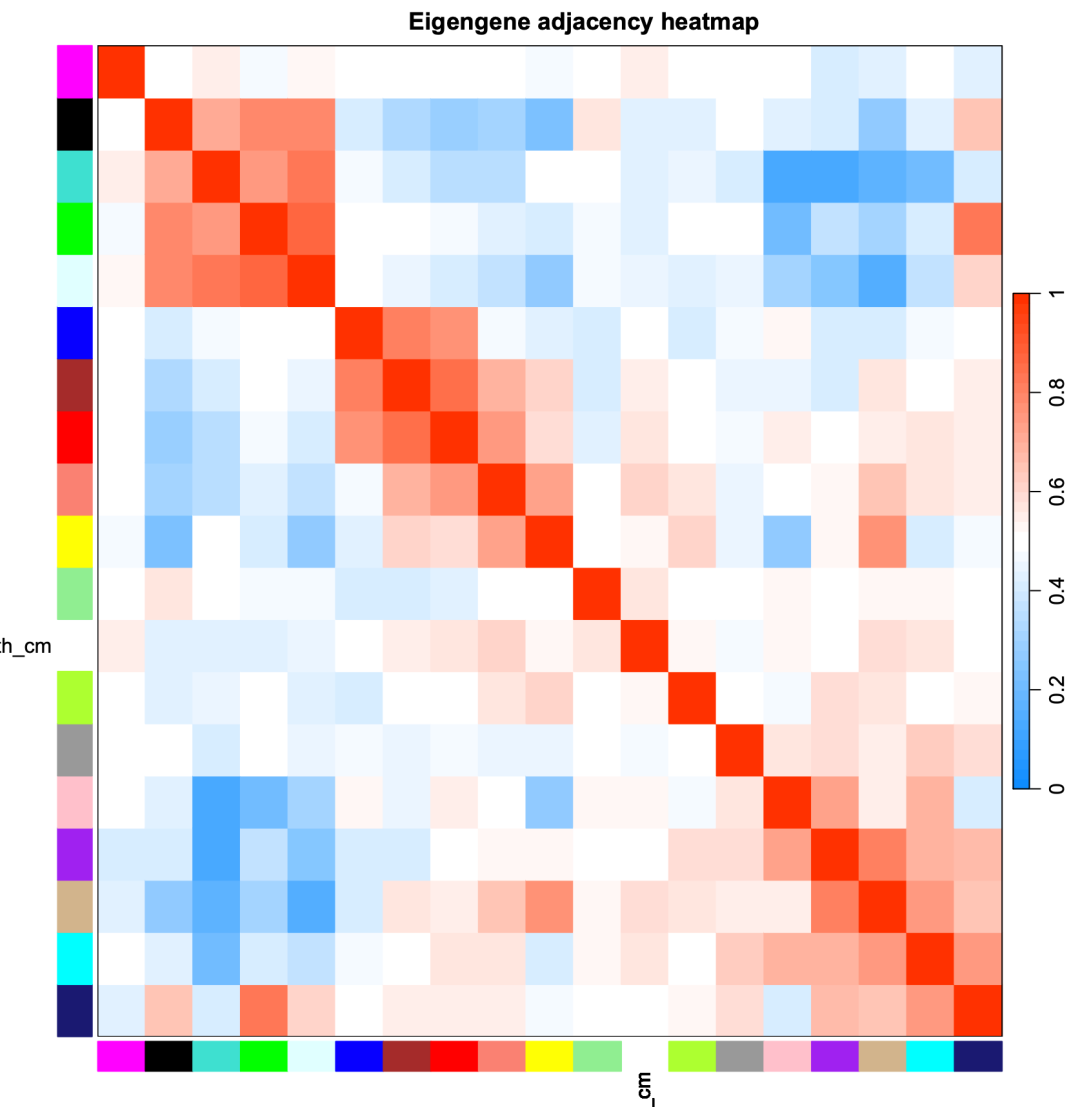
运行完毕,我们得到 4 个图:
- • 所有基因网络热图
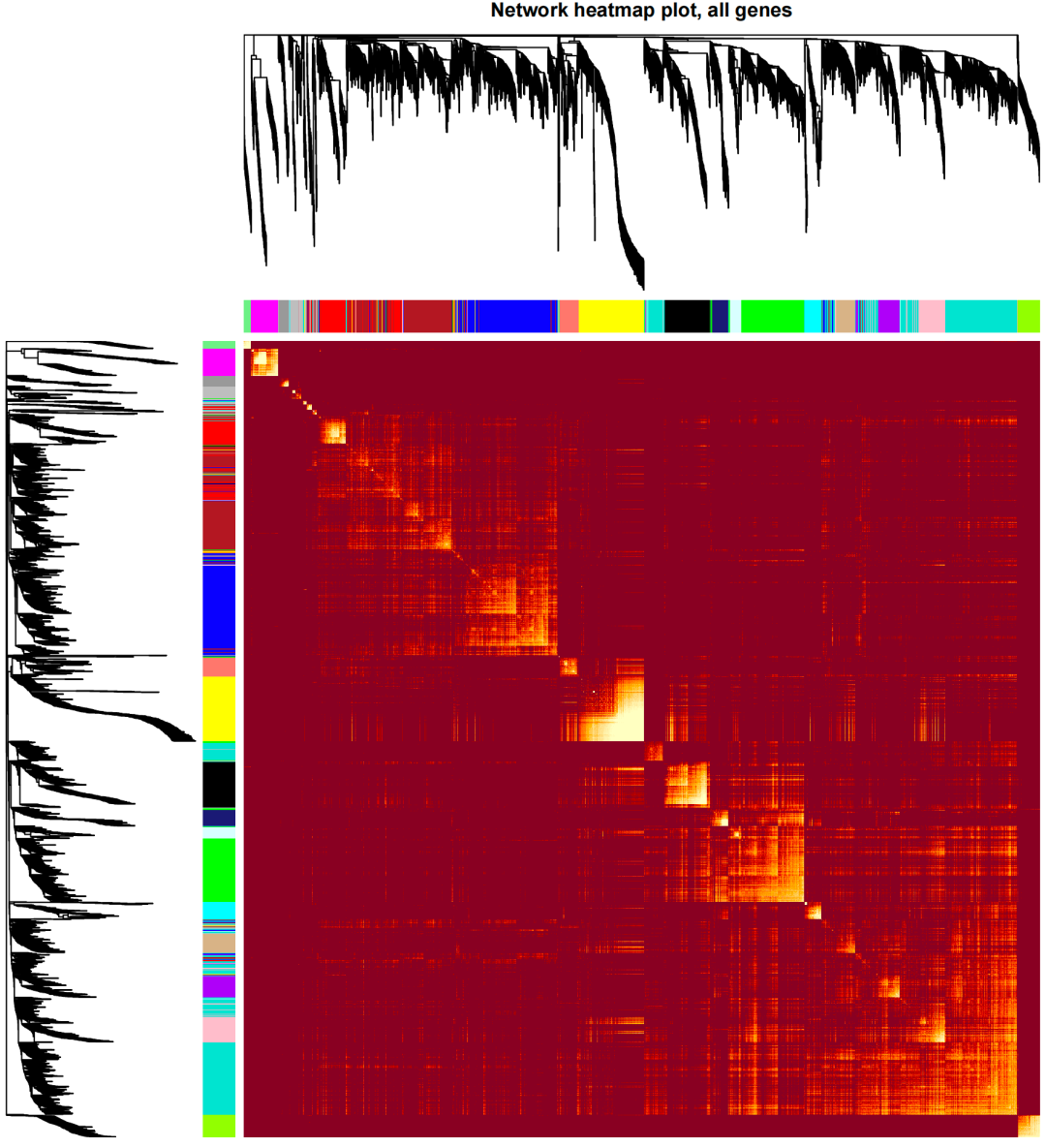
- • 模块聚类图和模块与特征相关性热图
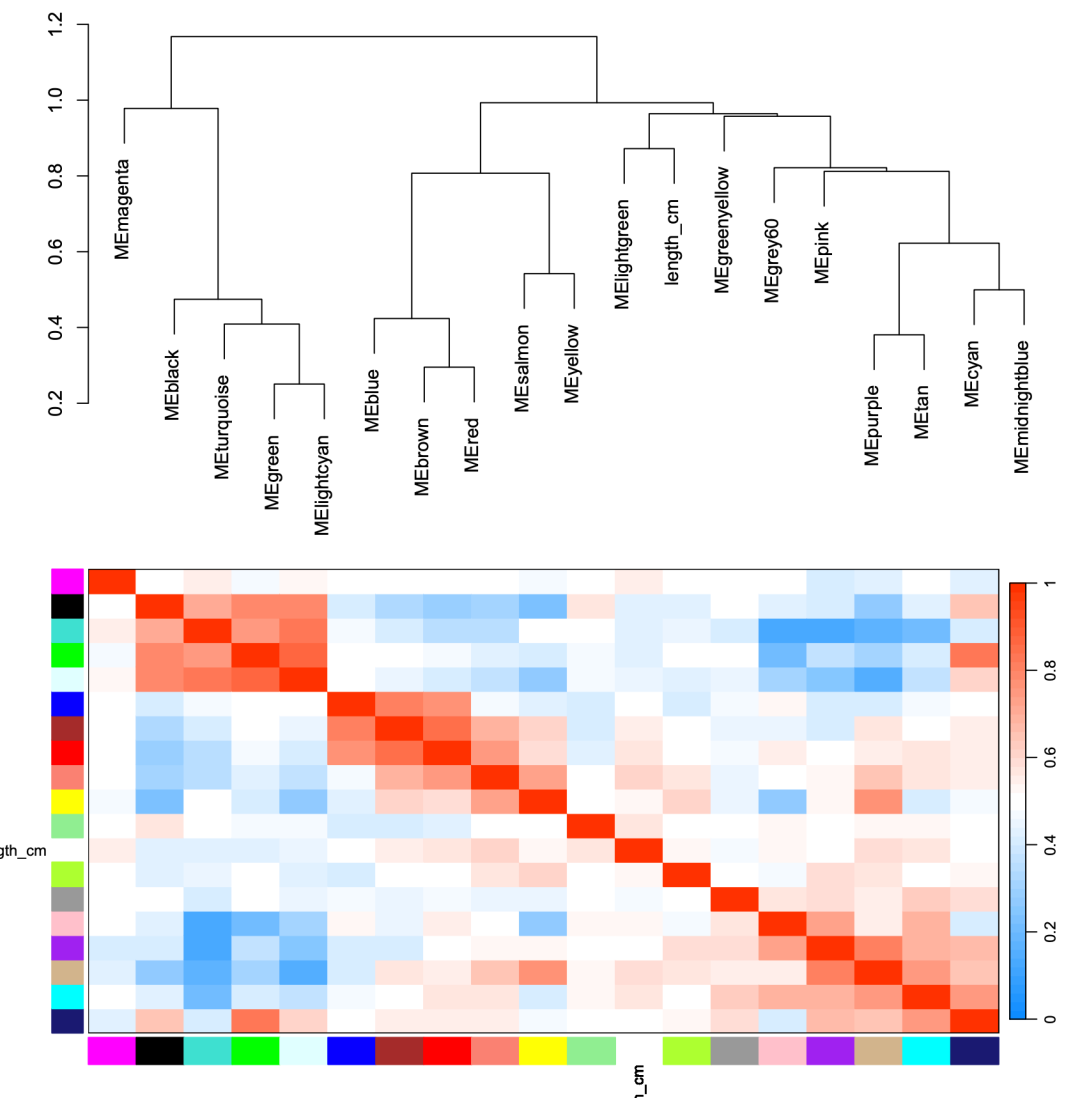
- • 模块聚类图
- • 模块与特征相关性热图
WGCNA STEP7
有时候,我们想挑选基因共表达网络中的一部分基因,用于 Cytoscape 作图。
参数设置:
- • Gene Expression Matrix: 过滤好的基因表达量矩阵
- • Gene List: 基因列表,文本文件,一行一个基因
- • Soft-threshold (Power) Source: 软阈值,可以手动填入,也可以用前面自动计算的

输出文件:
- • CytoscapeInput-edges.tsv
- • CytoscapeInput-nodes.tsv
这两个文件可以直接导入 Cytoscape,至此我们实现了挑选任意基因用于 Cytoscape 作图。
结语
通过本文,你应该学会了 WGCNA 分析,啃下了转录组分析的一块硬骨头。如果你对本文有什么疑问,或者建议,欢迎讨论。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-11-28,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号