生化小课 | 蛋白质序列有助于阐明地球上生命的历史(含蛋白质的结构:一级结构 小结)
生化小课 | 蛋白质序列有助于阐明地球上生命的历史(含蛋白质的结构:一级结构 小结)

生科/生技:生化书是我见过最厚的教材
没有之一
每周一堂
生化小课
—— 期末/考研 逢考必过——
蛋白质序列有助于阐明地球上生命的历史
表示蛋白质氨基酸序列的简单字母串包含惊人的丰富信息,通过将生物信息学工具应用于基因组和蛋白质序列数据,可以解锁这些信息。
每种蛋白质的功能都依赖于其三维结构,而三维结构又主要由其一级结构决定。因此,蛋白质序列传递的生化信息仅受限于我们对结构和功能原理的理解。不断发展的生物信息学工具使识别新蛋白质中的功能片段成为可能,也有助于确定它们的序列以及它们与数据库中已有蛋白质的结构关系。在不同层面的探究中,蛋白质序列开始告诉我们蛋白质是如何进化的,并最终告诉我们这个星球上的生命是如何进化的。
分子进化领域通常可以追溯到Emile Zuckerkandl和Linus Pauling,他们在20世纪60年代中期的工作推动了使用核苷酸和蛋白质序列来探索进化。前提看似简单。如果两个生物密切相关,那么它们的基因和蛋白质序列应该相似。随着两种生物之间进化距离的增加,序列的差异越来越大。这种方法的前景在 20世纪70年代开始实现,当时Carl Woese使用核糖体RNA序列将古细菌定义为一组不同于细菌和真核生物的生物体。如果我们能学会阅读遗传象形文字,基因组和蛋白质序列数据库中的信息就可以用来追溯生物历史。
进化并没有采取简单的线性路径。对于给定的蛋白质,蛋白质活性所必需的氨基酸残基在进化过程中是保守的。对功能不太重要的残基可能会随时间变化——也就是说,一种氨基酸可能会取代另一种——这些可变残基可以提供追踪进化的信息。一些蛋白质比其他蛋白质具有更多的可变氨基酸残基。由于这些和其他原因,不同的蛋白质以不同的速度进化。
追踪进化史的另一个复杂因素是一个基因或一组基因从一个生物体转移到另一个生物体的罕见情况,这一过程称为水平基因转移。转移的基因可能与它们在原始生物体中的来源基因相似,而这两种生物体中的大多数其他基因可能只是远缘相关。水平基因转移的一个例子是最近抗生素抗性基因在细菌种群中的快速传播。来自这些转移基因的蛋白质不会成为细菌进化研究的良好候选者,因为它们与其“宿主”生物仅共享非常有限的进化历史。
分子进化的研究通常侧重于密切相关的蛋白质家族。在大多数情况下,选择用于分析的家族在细胞代谢中具有基本功能,这些功能必须存在于最早的活细胞中,因此大大降低了它们最近通过水平基因转移引入的机会。例如,一种称为EF-1α(延伸因子 1α)的蛋白质参与所有真核生物的蛋白质合成。在细菌中发现了一种具有相同功能的类似蛋白质EF-Tu。序列和功能的相似性表明EF-1α和EF-Tu是具有共同祖先的蛋白质家族的成员。蛋白质家族的成员称为同源蛋白质或同源物。同源物的概念可以进一步细化。如果一个家族中的两种蛋白质(即两个同源物)存在于同一物种中,则它们被称为旁系同源物。来自不同物种的同源物称为直系同源物。追踪进化的过程包括首先识别合适的同源蛋白质家族,然后使用它们重建进化路径。
同源物是通过计算机程序识别的,计算机程序可以直接比较特定的蛋白质序列,或者可以搜索数据库,以识别在定义参数内匹配氨基酸的任何蛋白质。电子搜索过程可以被认为是将一个序列滑过另一个序列,直到找到一个匹配良好的部分。在此序列比对中,为两个序列相同的每个位置分配正分,在需要在一个序列或另一个序列中引入缺口以将其登记的任何位置引入负分。总分提供了比对质量的衡量标准(图 3-31)。该程序选择具有最佳分数的比对,最大限度地提高相同的氨基酸残基,同时尽量减少引入的差距。

发现相同的氨基酸往往不足以识别相关蛋白质,或者更重要的是,无法确定蛋白质在进化时间尺度上的密切程度。更有用的分析还考虑了取代氨基酸的化学性质。蛋白质家族中的许多氨基酸差异可能是保守的,也就是说,氨基酸残基被具有相似化学性质的残基所取代。例如,Glu残基可以在一个家族成员中替代另一个家族成员中的Asp残基;两种氨基酸都带负电荷。从逻辑上讲,这样的保守取代在序列比对中应该比非保守取代得到更高的分数——例如,用疏水性的Phe 残基取代Asp残基。
对于大多数寻找同源性和探索进化关系的努力,蛋白质序列优于不编码蛋白质或功能性 RNA 的核酸序列。对于具有四种不同类型残基的核酸,非同源序列的随机比对通常会产生至少 25% 的位置匹配。引入几个缺口通常可以将匹配残基的比例增加到40%或更多,并且不相关序列偶然比对的概率变得相当高。蛋白质中的20种不同的氨基酸残基大大降低了此类无信息机会比对的可能性。
用于生成序列比对的程序由测试比对可靠性的方法补充。一种常见的计算机化测试是打乱被比较的一种蛋白质的氨基酸序列以产生随机序列,然后指示程序将打乱的序列与另一个未打乱的序列对齐。分数分配给新的对齐方式,洗牌和对齐过程重复多次。在改组之前,原始比对的分数应明显高于随机比对生成的分数分布中的任何比对;这增加了序列比对已确定一对同源物的置信度。请注意,没有明显的比对分数并不一定意味着两种蛋白质之间不存在进化关系。正如我们将在第4章中看到的,三维结构相似性有时会揭示序列同源性已被时间抹去的进化关系。
为了使用蛋白质家族来探索进化,研究人员在尽可能广泛的生物体中识别出具有相似分子功能的家族成员。这些蛋白质家族中的序列差异允许根据生物的进化关系将生物分成几类。蛋白质序列的某些片段可能存在于一个分类群的生物体中,但不存在于其他群落中;这些片段可以用作它们所在组的签名序列。特征序列的一个例子是在所有古细菌和真核生物中的 EF-1α/EF-Tu蛋白的氨基末端附近插入12个氨基酸,但在细菌中则不然(图 3-32)。这个特殊的特征是许多生化线索之一,可以帮助建立真核生物和古细菌的进化相关性。

通过考虑多种蛋白质的序列,研究人员可以构建复杂的进化树。图3-33显示了10462种细菌的一个这样的树,基于细菌中普遍存在的120种蛋白质的序列。在图3-33中,线的自由端点被称为“外部节点”;每一个代表一个现存的物种,每一个都被如此标记。两条线合在一起的点,即“内部节点”,代表已灭绝的祖先物种。在大多数表示中(包括图3-33),连接节点的线的长度反映了将一个物种与另一个物种分离的所选蛋白质中的氨基酸取代。使用120种不同的蛋白质可以进行校准,并更准确地确定不同物种分化所需的时间。
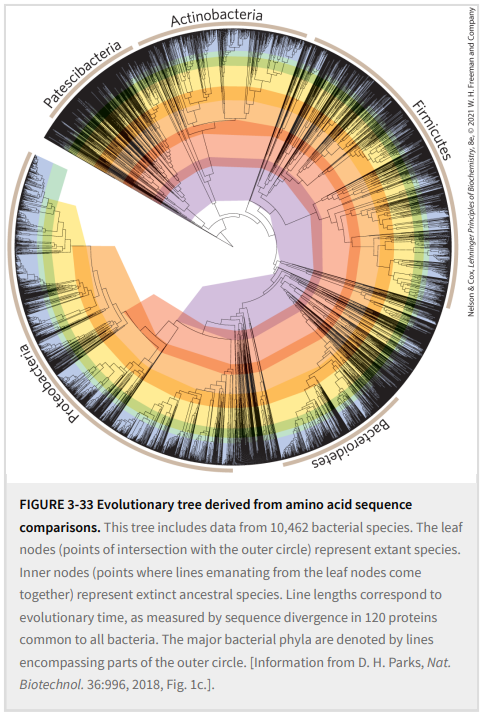
随着数据库中提供更多的序列信息,我们朝着生物学的核心目标之一迈进——创建详细的生命树,描述地球上每个生物体的进化和关系。这个故事正在进行中。提出和回答的问题是人类如何看待自己和周围世界的基础。
蛋白质的结构:一级结构
SUMMARY 3.4 The Structure ofProteins: Primary Structur
> 蛋白质功能的差异是由氨基酸组成和序列的差异造成的。特定氨基酸残基的化学性质往往对蛋白质的功能至关重要。
> 大多数氨基酸序列是从基因组序列和质谱法中推导出来的。从经典的蛋白质测序方法中获得的方法在蛋白质化学中仍然很重要。
> 短肽和多肽(最多约100个残基)可以进行化学合成。肽的建立是一种每次连接一个氨基酸残基的过程,同时与固定支撑物相连。
> 蛋白质序列是关于蛋白质结构和功能的丰富信息源。生物信息学可以分析同源蛋白质的氨基酸序列随时间的变化,以追踪地球生命的演化。
Principles of Biochemistry
本栏目信息及图片均来源于Lehninger Principles of Biochemistry 第八版,其中文字信息为英文原版的小编翻译/整理版,仅供学习交流使用,欢迎在留言区或私信听课君提供宝贵意见,如有侵权请联系删除。
Chapter Review部分未纳入翻译整理范围,如有需要建议参考原版图书该部分内容学习。
腾讯云开发者
