GEO2R更新后可以分析bulk RNAseq


随着生物学研究的不断发展,生物信息学变得越来越重要。研究人员需要有效的工具来分析、可视化和解释生物数据,这就为开发生物信息学工具提供了广泛的应用场景。而且最近几十年来,生物数据的生成速度呈指数级增长,需要不断更新和改进的工具来处理和解释这些数据。因此,有越来越多的需求来开发新的生物信息学工具或改进现有工具。具体到社会层面,产业应用,那就是随着个性化医疗的发展,需要针对患者的基因组数据开发个性化工具,这为工具开发人员提供了独特的机会。
要成为一个生物信息学领域的网页工具开发工程师,我们应该是着重加强以下能力和技能:
- 编程技能:学习编程语言,如Python、JavaScript、R、或Java等,以便能够编写、测试和维护网页应用程序。网页工具通常使用前端(如HTML、CSS、JavaScript)和后端(如Python、Node.js)技术。
- Web开发框架:熟悉常用的Web开发框架,如Django、Flask、React、Angular、或Vue.js等。这些框架可以加速网页工具的开发。
- 数据库管理:了解数据库管理系统,如MySQL、PostgreSQL、MongoDB等,以便存储和检索生物数据。
- 数据处理和分析:具备生物数据处理和分析的能力,包括数据清洗、统计分析、可视化等。这些技能对于将生物数据集成到网页工具中至关重要。
- 生物学知识:了解基本的生物学概念和术语,以便更好地理解用户需求和生物数据的含义。
- 数据隐私和安全:熟悉数据隐私和安全的最佳实践,以确保用户数据的安全和合法使用。
- 用户界面设计:具备用户界面设计的知识,以创建用户友好且易于导航的网页工具。
- 版本控制:使用版本控制工具,如Git,以便跟踪代码更改和合作开发。
- 协作和沟通:能够与团队合作,并有效沟通您的设计和开发决策。
- 持续学习:生物信息学领域和Web开发技术都在不断演进,因此持续学习和跟进最新的发展是必要的。
- 项目管理:学习项目管理方法,以便有效地计划、执行和交付网页工具项目。
- 测试和调试:熟悉测试和调试技术,以确保网页工具的稳定性和性能。
- 部署和维护:了解如何将网页工具部署到服务器上,并进行日常维护。
- 云计算:有云计算平台(如AWS、Azure、Google Cloud等)的基本知识,以便将网页工具部署到云上。
- 开源贡献:积极参与开源生物信息学项目,以建立声誉并学习与其他开发者合作的技能。
当然了,仅仅是做到这些还不够,我们还需要足够的资金支持,因为绝大部分网页工具的十几年如一日的维护推广和更新,也是不小的花销。相信大家应该是看到过无数的网页工具云平台如雨后春笋般出现和消失,这一点来说,由美国国立生物技术信息中心(NCBI)维护的一个公共数据库,用于存储和共享高通量基因表达数据的GEO(Gene Expression Omnibus)就是其中的佼佼者啦,它有一个在线分析工具GEO2R,用于比较两个或多个基因表达数据集,并识别在不同条件下表达显著差异的基因。用于快速的基因表达分析,研究人员可以使用它来比较不同实验条件下的基因表达差异,例如,疾病与对照组、不同治疗组之间的差异等。
GEO2R简介
GEO2R是由GEO数据库提供的一个交互式网络工具,允许用户比较GEO数据集中样本/组别间的差异,以确定在不同实验条件下差异表达的基因。GEO2R的结果显示为按p值排序的基因表,并作为图形图的集合,以帮助可视化差异表达基因和评估数据集质量。
GEO2R一般通过数据集下方的蓝色按钮进入并使用。
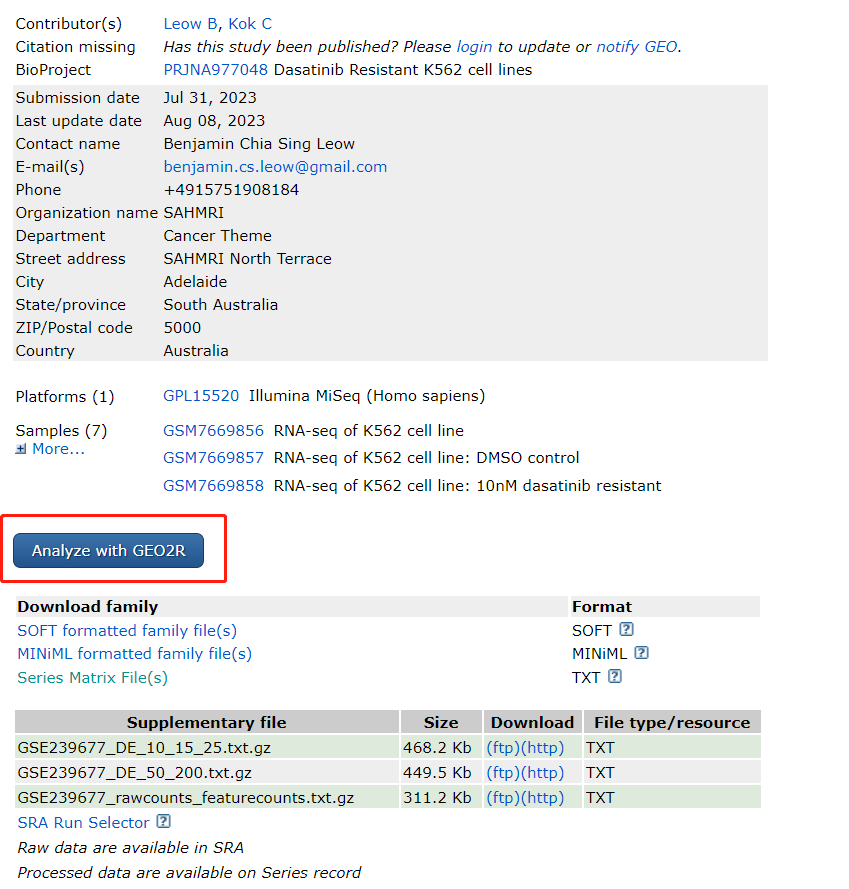
GEO2R入口
GEO2R的优缺点
GEO2R使用来自Bioconductor项目的各种R包,以最常用的芯片分析为例,其后台调用芯片分析的经典包Limma,通过数据清洗,差异分析等过程,最后获得用户可以直接使用的差异基因列表
优点:
1、无代码化。用户不需要任何编程基础,可以完全通过鼠标“点点点”的方式进行样品的分组和差异分析,并可以直接获得可视化结果和差异基因列表。
2、用户无需预处理数据。芯片的差异分析之前需要对芯片信号进行定量,GEO会对芯片数据的原始数据进行信号定量分析并形成矩阵,用于GEO2R调用,因此用户无需进行预处理数据,降低使用门槛。
3、输出结果内容全面。输出结果中不仅包含差异基因列表,还包括常见的样本count分布图、质控图、火山图、PCA图等,甚至每个差异基因在网页上点看还能看到分组的表达量图。虽然比较丑但贵在实用!
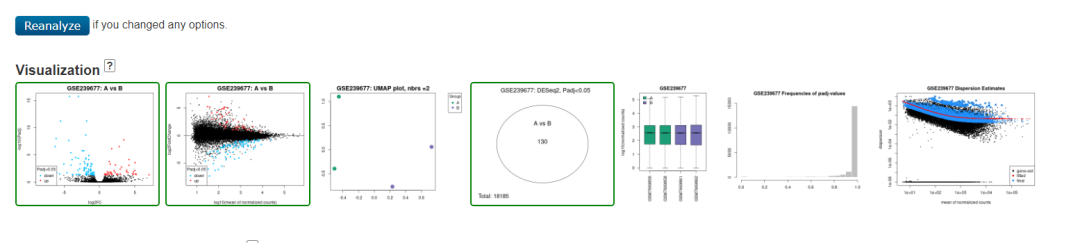
GEO2R输出结果
缺点:
1、目前芯片用得越来越少,RNAseq越来越普及,本次更新前不支持RNAseq数据一直是弊病,限制它的使用。这次更新终于解决这个问题了!
2、无法做后续的富集分析。好在可以通过metascape或DAVID等在线工具解决,不是什么大问题。ps:差异分析又快又好,还免费,要啥自行车?
GEO2R beta版更新
详细内容见:https://www.ncbi.nlm.nih.gov/geo/info/geo2r.html
1、亮点
主要增加了对RNAseq数据分析的支持。目前GEO2R支持使用DESeq2对GEO及SRA库中的数据进行差异分析,输入文件是NCBI-computed raw count matrices。
2、NCBI-computed raw count matrices
GEO对于研究者上传到SRA的转录组数据重新进行比对和定量获取NCBI-computed raw count matrices,流程是hisat2+featurecounts(subread包),并且提供了count矩阵的下载方式的API,格式参考下面的示例:https://www.ncbi.nlm.nih.gov/geo/download/?type=rnaseq_counts&acc=GSE164073&format=file&file=GSE164073_raw_counts_GRCh38.p13_NCBI.tsv.gz(这里把GSE编号和替换成自己的就可以了)用户可以通过该API直接获得表达矩阵,可以用于后续的分析(本地用R语言自己处理),也可以不下载,直接用GEO2R在线处理。
NCBI-computed raw count matrices目前暂时只做了人和小鼠的,其中人的数据已经释放,小鼠的数据会在2023年秋季释放,因此目前暂时只能用GEO2R分析人的数据。
目前单细胞的表达矩阵不在更新范围内
3、NCBI-generated data的其它说明
既然有了count矩阵,很多小伙伴就问了,有没有FPKM和TPM矩阵呢?GEO团队早早早就考虑过了,本次更新除了count矩阵外,还顺便释放了FPKM和TPM矩阵,以及基因注释文件。这些文件可以通过API获取,示例如下:https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE164073。这里把GSE编号替换成自己的就可以了。
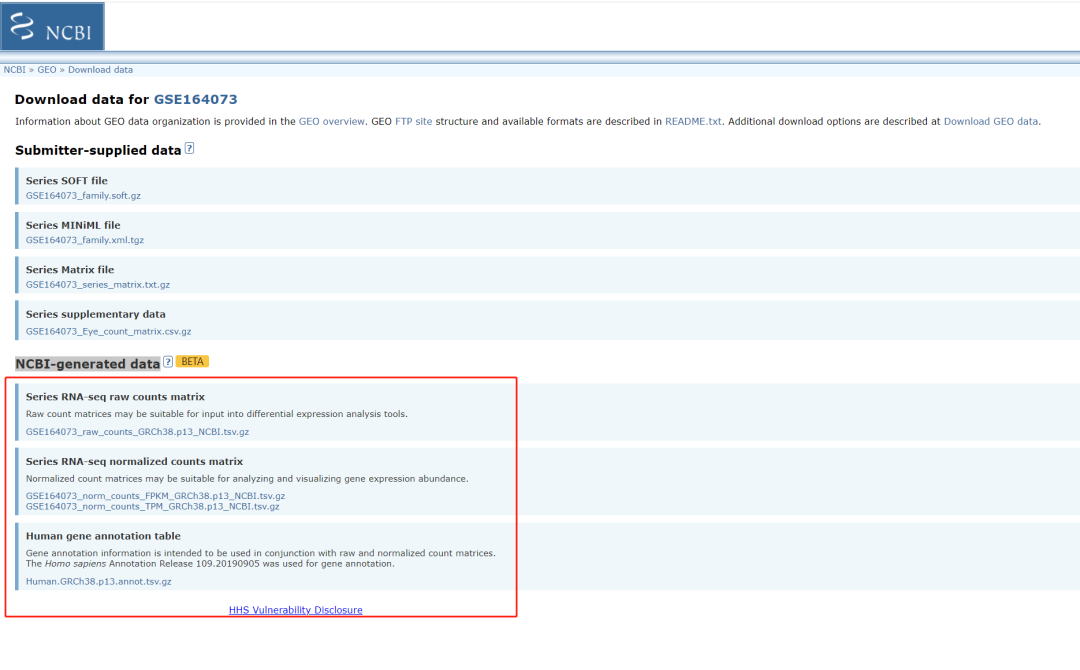
获取所有的NCBI-generated data
GEO2R如何分析RNAseq数据?
GEO2R分析RNAseq的方法与分析芯片的方法只能说是一模一样。具体见生信技能树的帖子:https://cloud.tencent.com/developer/article/1625367,也可以直接搜,一大堆。
如果仍然搞不明白,可以参考GEO团队制作的演示视频:https://youtu.be/9RyWjzSnaE0
对于GEO2R输出结果的解读,可以参考说明文件,写得巨详细:https://www.ncbi.nlm.nih.gov/geo/info/geo2r.html
大家很容易拿到绝大部分转录组测序数据集对应的gse数据集的表达量矩阵:
比如 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130437 就是一个简简单单的转录组测序, Two cell lines (MDA-MB231 and MCF7) with two treatments ,那就是4X3=12个样品啦 :
- https://www.ncbi.nlm.nih.gov/geo/download/?type=rnaseq_counts&acc=GSE130437&format=file&file=GSE130437_raw_counts_GRCh38.p13_NCBI.tsv.gz
值得注意的是,如果一个gse数据集里面不仅仅是有转录组测序, 还有其它测序,比如 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133399,里面就是 T cells in lung treated with Aspergillus antigen are assessed by RNA-seq and ATAC-Seq.
这样的话,同样的方法就拿不到矩阵的哦:
- https://www.ncbi.nlm.nih.gov/geo/download/?type=rnaseq_counts&acc=GSE130437&format=file&file=GSE130437_raw_counts_GRCh38.p13_NCBI.tsv.gz
会给你如下所示的报错信息:
There was a problem executing your request. If the problem persists, please write to geo@ncbi.nlm.nih.gov, describing in detail what you were trying to do and quoting the following message:
2023-09-09T11:38:36.633258 2F3402163625F383_22830SID
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2023-09-10 16:37,如有侵权请联系 cloudcommunity@tencent.com 删除
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号