生信技能树-day19 转录组下游分析-标准化、聚类、差异分析

生信技能树-day19 转录组下游分析-标准化、聚类、差异分析

生信菜鸟团
发布于 2024-06-25 20:21:15
发布于 2024-06-25 20:21:15
从我们生信技能树历年的几千个马拉松授课学员里面募集了一些优秀的创作者,某种意义来说是传承了我们生信技能树的知识整理和分享的思想!
准备工作
数据标准化
标准化前需要进行数据预处理
过滤低表达的基因,并检查是否有异常样本
以下是常见的几种过滤方式(过滤的标准都可以自己调整)
- 在至少在75%的样本中都表达的基因(表达是指在某个样本中count值>0)
- 过滤平均值count<10的基因
- 过滤平均cpm <10 的基因
为什么做标准化?
标准化的主要目的是去除测序数据的测序深度和基因长度。
• 测序深度:每个样本的测序深度(产生的数据量)不完全一样,同一条件下,测序深度越深,基因表达的read读数越多。
• 基因长度:同一条件下,不同的基因长度产生不对等的read读数,基因越长,该基因的read读数越高。视基因类型而定,比如microRNA稳定在20+长度,则不用针对长度进行标准化
FPKM值
Fragments per kilobase per Million mapped reads 片段数量per千碱基长度per百万比对上的reads
FPKM = cDNA Fragments/Mapped Fragments(Millions)×Transcript Length(kb)
• cDNA Fragments:比对到某一转录本上的片段数目,即双端Reads数目,针对双端数据;
• Mapped Fragments (Millions):比对到转录本上的片段总数,以百万为单位,即10^6;
• Transcript Length(kb):转录本长度,kb为单位,即10^3。
RPM值
RPM:Reads of exon model per Million mapped reads
RPM=total exon reads / mapped reads (Millions)
RPM方法:标准化了测序深度的影响,但没有考虑转录本的长度的影响。
RPM适合于产生的read读数不受基因长度影响的测序方法,比如miRNAseq测序,miRNA的长度一般在18~32个碱基之间。
TPM值
TPM,Transcripts Per Kilobase Million
TPMi=(Ni/Li)*1000000/sum(Ni/Li+……..+ Nm/Lm)
和FPRM值一样都矫正了深度和长度,但TPM先对每个基因的read数用基因的长度进行校正,之后再用校正后的这个基因read数(Ni/Li)与校正后的这个样本的所有read数(sum(Ni/Li+……..+ Nm/Lm))求商,最后再乘以10^6。最后每个样本的TPM加和是相等的
Ni:mapping到基因i上的read数;
Li:基因i的外显子长度的总和
各种值的使用场合
- 差异表达分析:原始count值,算法输入要求(针对二代测序差异分析算法,算法内部一般有标化方法。)
2.标化后的值:基因表达值在样本与样本之间具有可比性。比如PCA分析,样本表达总体分布,生存分析,热图绘制,相关性分析等
实际代码
rm(list = ls())
library(stringr)
## ====================1.读取数据
# 读取raw count表达矩阵
rawcount <- read.table("data/raw_counts.txt",row.names = 1,
sep = "\t", header = T)
colnames(rawcount)
## [1] "SRR1039508" "SRR1039509" "SRR1039512" "SRR1039513" "SRR1039516"
## [6] "SRR1039517" "SRR1039520" "SRR1039521"
# 查看表达谱
rawcount[1:4,1:4]
## SRR1039508 SRR1039509 SRR1039512 SRR1039513
## ENSG00000000003 679 448 873 408
## ENSG00000000005 0 0 0 0
## ENSG00000000419 467 515 621 365
## ENSG00000000457 260 211 263 164
# 去除前的基因表达矩阵情况
dim(rawcount)
## [1] 64102 8
# 获取分组信息
# 自己做一个分组信息的表格,第一列是样本,第二列是分组信息
group <- read.table("data/group.txt",header = T,sep = "\t", quote = "\"")
group
## run_accession sample_title
## 1 SRR1039508 untreated
## 2 SRR1039509 Dex
## 3 SRR1039512 untreated
## 4 SRR1039513 Dex
## 5 SRR1039516 untreated
## 6 SRR1039517 Dex
## 7 SRR1039520 untreated
## 8 SRR1039521 Dex
## =================== 2.表达矩阵预处理
# 过滤低表达基因
# rawcount>0判断后生成一个和rawcount表达矩阵对应的逻辑向量
# rawSums统计每个基因在每个样本中生成的逻辑向量的和,F为0,T为1,则为每个基因在多少个样本中有表达
# ncol指样本的数量,0.75*ncol(rawcount)指至少在75%的样本中有表达,再floor向下取整
keep <- rowSums(rawcount>0) >= floor(0.75*ncol(rawcount))
table(keep)
## keep
## FALSE TRUE
## 42172 21930
# 生成一个判断满足在75%的样本中有表达的逻辑向量矩阵
# 如果按平均值count<10,则需要改成rowMeans(rawcount)>10,更为严格
# 按keep逻辑向量矩阵取rawcount的子集,生成一个过滤后的矩阵filter_count
filter_count <- rawcount[keep,]
# 简单看一下确认是对的
filter_count[1:4,1:4]
## SRR1039508 SRR1039509 SRR1039512 SRR1039513
## ENSG00000000003 679 448 873 408
## ENSG00000000419 467 515 621 365
## ENSG00000000457 260 211 263 164
## ENSG00000000460 60 55 40 35
dim(filter_count)
## [1] 21930 8
# 此处选择使用cpm值进行标准化
# 加载edgeR包计算counts per million(cpm) 表达矩阵
library(edgeR)
express_cpm <- cpm(filter_count)
express_cpm[1:6,1:6]
## SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516
## ENSG00000000003 32.908572 23.824498 34.449640 26.91285 46.559683
## ENSG00000000419 22.633731 27.387536 24.505414 24.07645 24.016287
## ENSG00000000457 12.601221 11.220913 10.378299 10.81791 10.023833
## ENSG00000000460 2.907974 2.924883 1.578449 2.30870 3.191261
## ENSG00000000971 157.563723 195.648051 243.751919 280.47412 274.980344
## ENSG00000001036 69.452112 56.476823 68.386284 58.11329 58.260975
## SRR1039517
## ENSG00000000003 33.982142
## ENSG00000000419 25.932886
## ENSG00000000457 10.743161
## ENSG00000000460 2.044771
## ENSG00000000971 357.899793
## ENSG00000001036 46.705160
# 保存表达矩阵和分组结果
save(filter_count, express_cpm, group, file = "data/Step01-airwayData.Rdata")
异常样本与重复性检测
why需要这一步?
- 检查组内样本的重复性是否好
- 检查组间样本是否可以较好的分开
步骤
- 样本表达水平总体分布图
- 样本相关性检验
- PCA分析
- 层次聚类分析
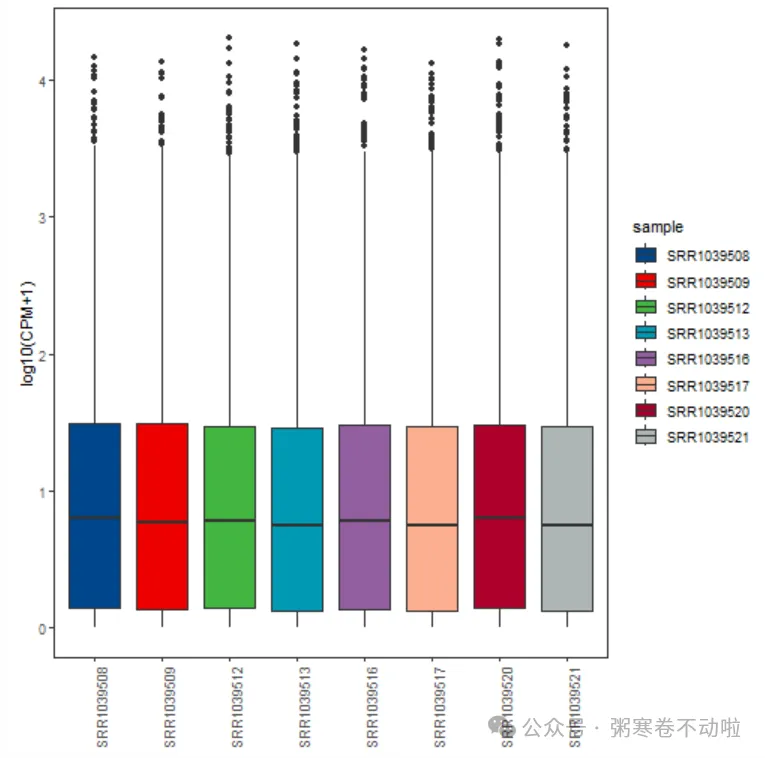
样本表达总体分布图
使用箱线图/小提琴图/概率密度分布图,观察:
- 单个样本的数据分布特点
- 多个样本之间的差异
rm(list = ls())
options(stringsAsFactors = F)
# 加载包,设置绘图参数
library(ggplot2)
library(ggsci)
library(tidyverse)
# 设置一个喜欢的主题
mythe <- theme_bw() + theme(panel.grid.major=element_blank(),
panel.grid.minor=element_blank())
# 加载原始表达的数据
lname <- load(file = "data/Step01-airwayData.Rdata")
lname
## [1] "filter_count" "express_cpm" "group"
# cpm值的矩阵取1og10后+1,生成用于比较画图的表达矩阵
exprSet <- log10(as.matrix(express_cpm)+1)
exprSet[1:6,1:6]
## SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516
## ENSG00000000003 1.5303095 1.3948805 1.5496118 1.4458042 1.6772390
## ENSG00000000419 1.3735323 1.4531277 1.4066324 1.3992660 1.3982228
## ENSG00000000457 1.1335779 1.0871037 1.0560774 1.0725407 1.0423326
## ENSG00000000460 0.5919517 0.5938267 0.4113585 0.5196574 0.6223447
## ENSG00000000971 2.2002038 2.2936896 2.3887261 2.4494385 2.4408782
## ENSG00000001036 1.8478940 1.7594928 1.8412736 1.7716851 1.7727688
## SRR1039517
## ENSG00000000003 1.5438464
## ENSG00000000419 1.4302829
## ENSG00000000457 1.0697850
## ENSG00000000460 0.4835546
## ENSG00000000971 2.5549732
## ENSG00000001036 1.6785654
## 1.样本表达总体分布-箱式图
# 构造绘图数据:宽数据转为长数据
# 把所有数据都放到一列中,列名的值的新列名为“sample”,值的新列名为“expression”,基因编号丢弃
data <- exprSet %>%
as.data.frame() %>%
pivot_longer(cols = everything(), names_to = "sample",values_to = "expression")
head(data)
## # A tibble: 6 × 2
## sample expression
## <chr> <dbl>
## 1 SRR1039508 1.53
## 2 SRR1039509 1.39
## 3 SRR1039512 1.55
## 4 SRR1039513 1.45
## 5 SRR1039516 1.68
## 6 SRR1039517 1.54
# 画箱式图
p <- ggplot(data = data, aes(x=sample,y=expression,fill=sample))
p1 <- p + geom_boxplot() +
mythe+ theme(axis.text.x = element_text(angle = 90)) +
xlab(NULL) + ylab("log10(CPM+1)") + scale_fill_lancet()
p1
# 保存图片
png(file = "result/1.sample_boxplot.png",width = 800, height = 900,res=150)
print(p1)
dev.off()
## png
## 2
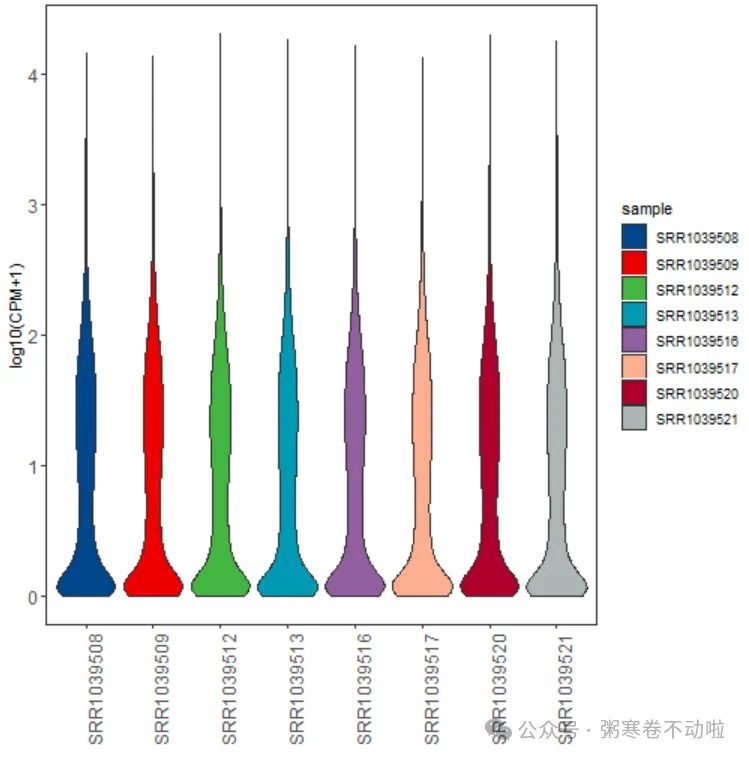
## 2.样本表达总体分布-小提琴图
p2 <- p + geom_violin() + mythe +
theme(axis.text = element_text(size = 12),
axis.text.x = element_text(angle = 90)) +
xlab(NULL) + ylab("log10(CPM+1)")+scale_fill_lancet()
p2
# 保存图片
png(file = "result/1.sample_violin.png",width = 800, height = 900,res=150)
print(p2)
dev.off()
## png
## 2
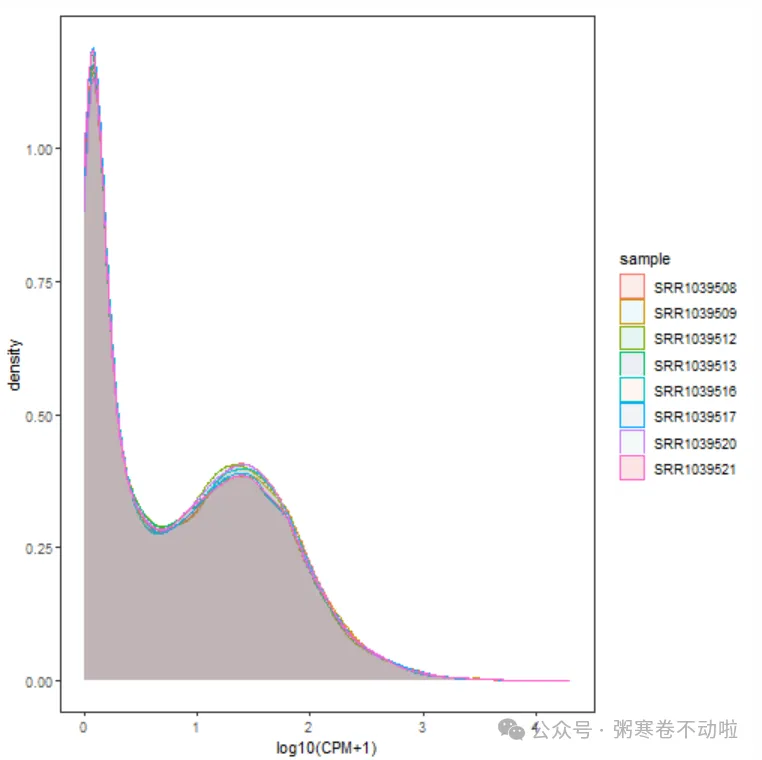
## 3.样本表达总体分布-概率密度分布图
m <- ggplot(data=data, aes(x=expression))
p3 <- m + geom_density(aes(fill=sample, colour=sample),alpha = 0.1) +
xlab("log10(CPM+1)") + mythe +scale_fill_npg()
p3
# 保存图片
png(file = "result/1.sample_density.png",width = 800, height = 700, res=150)
print(p3)
dev.off()
## png
## 2
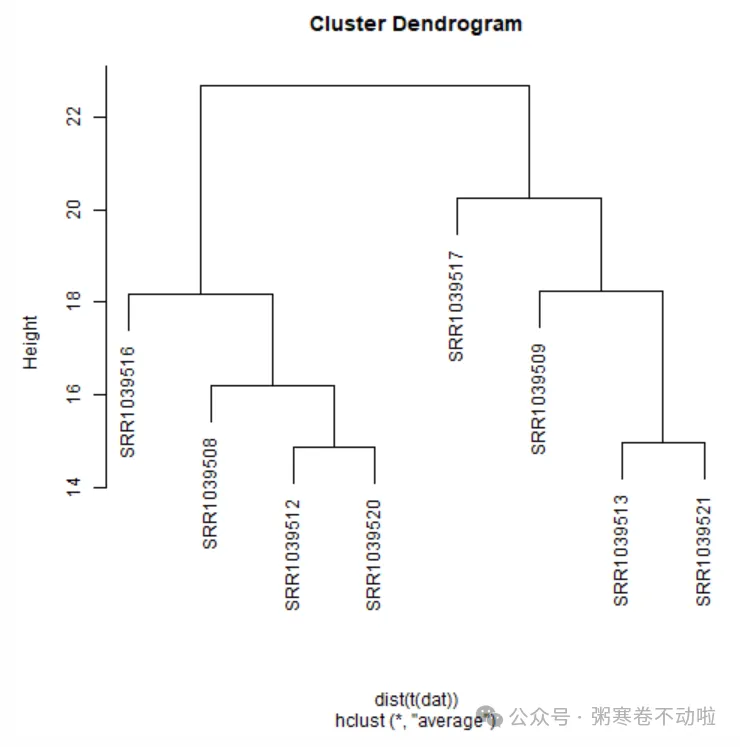
层次聚类
层次聚类树
通过计算点与点之间的空间距离对样本进行类别划分
# 清空环境变量
rm(list = ls())
options(stringsAsFactors = F)
library(FactoMineR)
library(factoextra)
library(corrplot)
library(pheatmap)
library(tidyverse)
# 加载数据并检查
lname <- load(file = 'data/Step01-airwayData.Rdata')
lname
## [1] "filter_count" "express_cpm" "group"
## 1.样本之间的相关性-层次聚类树----
# 取log
dat <- log10(express_cpm+1)
dat[1:4,1:4]
## SRR1039508 SRR1039509 SRR1039512 SRR1039513
## ENSG00000000003 1.5303095 1.3948805 1.5496118 1.4458042
## ENSG00000000419 1.3735323 1.4531277 1.4066324 1.3992660
## ENSG00000000457 1.1335779 1.0871037 1.0560774 1.0725407
## ENSG00000000460 0.5919517 0.5938267 0.4113585 0.5196574
dim(dat)
## [1] 21930 8
# 画层次聚类树
# t进行转置,dist计算样本和样本之间的距离,有不同的距离,如曼哈顿距离,欧氏距离等,计算方法不同
# hclust得到的一个矩阵称为下三角矩阵,值为样本和样本之间的相似度(距离),里面还有如计算方法等信息
sampleTree <- hclust(dist(t(dat)), method = "average")
plot(sampleTree)
# 提取样本聚类信息
temp <- as.data.frame(cutree(sampleTree,k = 2)) %>%
rownames_to_column(var="sample")
temp1 <- merge(temp,group,by.x = "sample",by.y="run_accession")
table(temp1$`cutree(sampleTree, k = 2)`,temp1$sample_title)
##
## Dex untreated
## 1 0 4
## 2 4 0
# 保存结果
pdf(file = "result/2.sample_Treeplot.pdf",width = 7,height = 6)
plot(sampleTree)
dev.off()
## png
## 2
PCA图
通过提取样本的综合特征,即主成分(第一主成分,第二主成分...)来对样本进行分类
## 2.样本之间的相关性-PCA----
# 第一步,数据预处理
dat <- log10(express_cpm+1)
dat[1:4,1:4]
## SRR1039508 SRR1039509 SRR1039512 SRR1039513
## ENSG00000000003 1.5303095 1.3948805 1.5496118 1.4458042
## ENSG00000000419 1.3735323 1.4531277 1.4066324 1.3992660
## ENSG00000000457 1.1335779 1.0871037 1.0560774 1.0725407
## ENSG00000000460 0.5919517 0.5938267 0.4113585 0.5196574
dat <- as.data.frame(t(dat))
dat_pca <- PCA(dat, graph = FALSE)
group_list <- group[match(group$run_accession,rownames(dat)), 2]
group_list
## [1] "untreated" "Dex" "untreated" "Dex" "untreated" "Dex"
## [7] "untreated" "Dex"
# geom.ind: point显示点,text显示文字
# palette: 用不同颜色表示分组
# addEllipses: 是否圈起来
mythe <- theme_bw() +
theme(panel.grid.major=element_blank(),panel.grid.minor=element_blank()) +
theme(plot.title = element_text(hjust = 0.5))
p <- fviz_pca_ind(dat_pca,
geom.ind = "point", #point
col.ind = group_list,
palette = c("#00AFBB", "#E7B800"),
addEllipses = T,
legend.title = "Groups") + mythe
p
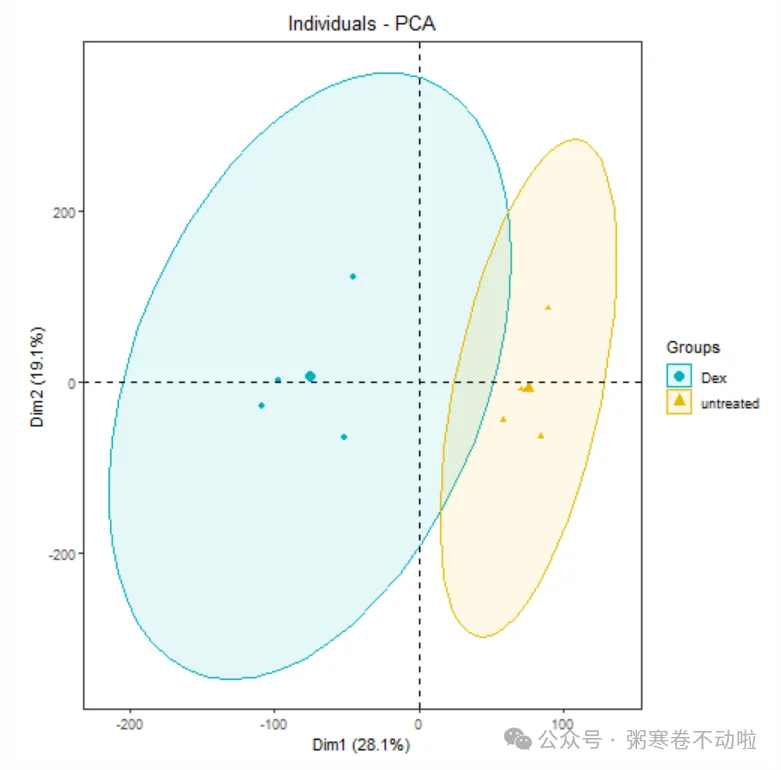
# 保存结果
pdf(file = "result/2.sample_PCA.pdf",width = 6.5,height = 6)
plot(p)
dev.off()
## png
## 2
相关性分析
通过计算样本与样本之间的相关性系数来对样本进行分类,相关性系数可以是pearson, spearman, kendall,不同的方法要求的数据预处理有所不同,如pearson要取log
## 3.样本之间的相关性-cor----
# 选择差异变化大的基因算样本相关性
exprSet <- express_cpm
exprSet = exprSet[names(sort(apply(exprSet, 1, mad),decreasing = T)[1:800]),]
dim(exprSet)
## [1] 800 8
# 计算相关性
M <- cor(exprSet,method = "spearman")
M
## SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517
## SRR1039508 1.0000000 0.8626996 0.9516678 0.7734420 0.9366474 0.7944256
## SRR1039509 0.8626996 1.0000000 0.8400846 0.9101584 0.8293501 0.8922465
## SRR1039512 0.9516678 0.8400846 1.0000000 0.8477138 0.9312893 0.8369531
## SRR1039513 0.7734420 0.9101584 0.8477138 1.0000000 0.7827427 0.9110153
## SRR1039516 0.9366474 0.8293501 0.9312893 0.7827427 1.0000000 0.8727728
## SRR1039517 0.7944256 0.8922465 0.8369531 0.9110153 0.8727728 1.0000000
## SRR1039520 0.9539928 0.8248631 0.9657655 0.8175386 0.9248946 0.8064216
## SRR1039521 0.7715402 0.9123616 0.8176518 0.9671464 0.7749063 0.8952126
## SRR1039520 SRR1039521
## SRR1039508 0.9539928 0.7715402
## SRR1039509 0.8248631 0.9123616
## SRR1039512 0.9657655 0.8176518
## SRR1039513 0.8175386 0.9671464
## SRR1039516 0.9248946 0.7749063
## SRR1039517 0.8064216 0.8952126
## SRR1039520 1.0000000 0.8321493
## SRR1039521 0.8321493 1.0000000
# 构造注释条
anno <- data.frame(group=group$sample_title,row.names = group$run_accession )
# 保存结果
pheatmap(M,display_numbers = T,
annotation_col = anno,
fontsize = 10,cellheight = 30,
cellwidth = 30,cluster_rows = T,
cluster_cols = T,
filename = "result/2.sample_Cor.pdf",width = 7.5,height = 7)
差异表达分析
步骤:
- 创建设计矩阵和对比
- 构建edgeR的DGEList对象,并归一化,拟合模型
- 提取分析结果并筛选显著差异基因
rm(list = ls())
options(stringsAsFactors = F)
# 加载包
library(edgeR)
library(ggplot2)
# 读取基因表达矩阵信息并查看分组信息和表达矩阵数据
lname <- load(file = "data/Step01-airwayData.Rdata")
lname
## [1] "filter_count" "express_cpm" "group"
# 表达谱
filter_count[1:4,1:4]
## SRR1039508 SRR1039509 SRR1039512 SRR1039513
## ENSG00000000003 679 448 873 408
## ENSG00000000419 467 515 621 365
## ENSG00000000457 260 211 263 164
## ENSG00000000460 60 55 40 35
# 分组信息
group_list <- group[match(colnames(filter_count),group$run_accession),2]
group_list
## [1] "untreated" "Dex" "untreated" "Dex" "untreated" "Dex"
## [7] "untreated" "Dex"
# treat vs control
comp <- unlist(strsplit("Dex_vs_untreated",split = "_vs_"))
group_list <- factor(group_list,levels = comp)
group_list
## [1] untreated Dex untreated Dex untreated Dex untreated
## [8] Dex
## Levels: Dex untreated
table(group_list)
## group_list
## Dex untreated
## 4 4
# 构建线性模型。0代表x线性模型的截距为0
design <- model.matrix(~0+group_list)
rownames(design) <- colnames(filter_count)
colnames(design) <- levels(factor(group_list))
design
## Dex untreated
## SRR1039508 0 1
## SRR1039509 1 0
## SRR1039512 0 1
## SRR1039513 1 0
## SRR1039516 0 1
## SRR1039517 1 0
## SRR1039520 0 1
## SRR1039521 1 0
## attr(,"assign")
## [1] 1 1
## attr(,"contrasts")
## attr(,"contrasts")$group_list
## [1] "contr.treatment"
# 构建edgeR的DGEList对象
DEG <- DGEList(counts=filter_count,
group=factor(group_list))
# 归一化基因表达分布
DEG <- calcNormFactors(DEG)
# 计算线性模型的参数
DEG <- estimateGLMCommonDisp(DEG,design)
DEG <- estimateGLMTrendedDisp(DEG, design)
DEG <- estimateGLMTagwiseDisp(DEG, design)
# 拟合线性模型
fit <- glmFit(DEG, design)
# 进行差异分析,计算LRT值
lrt <- glmLRT(fit, contrast=c(1,-1))
# 提取过滤差异分析结果
# 其中LR为上面计算的LRT值,FDR值为矫正后的P值
DEG_edgeR <- as.data.frame(topTags(lrt, n=nrow(DEG)))
head(DEG_edgeR)
## logFC logCPM LR PValue FDR
## ENSG00000152583 4.595657 5.542809 390.4758 6.520763e-87 1.430003e-82
## ENSG00000109906 7.149822 4.171049 244.7369 3.646653e-55 3.998555e-51
## ENSG00000179094 3.178430 5.182958 211.8321 5.472245e-48 4.000211e-44
## ENSG00000189221 3.298028 6.772968 207.5628 4.673127e-47 2.562042e-43
## ENSG00000120129 2.942776 7.314826 189.1498 4.874192e-43 2.137821e-39
## ENSG00000096060 3.930266 6.903020 186.7344 1.641237e-42 5.998721e-39
# 筛选上下调,设定阈值
# 筛选显著 差异 表达基因
# 差异:通过设定Fold Change的阈值
# 差异:通过设定p值的阈值
fc_cutoff <- 1.5
pvalue <- 0.05
# 先把默认值都设置为normal
DEG_edgeR$regulated <- "normal"
# which返回满足两个条件的位置信息,取交集
loc_up <- intersect(which( DEG_edgeR$logFC > log2(fc_cutoff) ),
which( DEG_edgeR$PValue < pvalue) )
loc_down <- intersect(which(DEG_edgeR$logFC < (-log2(fc_cutoff))),
which(DEG_edgeR$PValue<pvalue))
# 把对应位置的regulated参数设置为up和down
DEG_edgeR$regulated[loc_up] <- "up"
DEG_edgeR$regulated[loc_down] <- "down"
table(DEG_edgeR$regulated)
##
## down normal up
## 1396 19252 1282
## 将Ensembl ID转换为gene symbol
# 方法1:使用包
library(org.Hs.eg.db)
keytypes(org.Hs.eg.db)
## [1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT"
## [5] "ENSEMBLTRANS" "ENTREZID" "ENZYME" "EVIDENCE"
## [9] "EVIDENCEALL" "GENENAME" "GENETYPE" "GO"
## [13] "GOALL" "IPI" "MAP" "OMIM"
## [17] "ONTOLOGY" "ONTOLOGYALL" "PATH" "PFAM"
## [21] "PMID" "PROSITE" "REFSEQ" "SYMBOL"
## [25] "UCSCKG" "UNIPROT"
library(clusterProfiler)
id2symbol <- bitr(rownames(DEG_edgeR),
fromType = "ENSEMBL",
toType = "SYMBOL",
OrgDb = org.Hs.eg.db)
head(id2symbol)
## ENSEMBL SYMBOL
## 1 ENSG00000152583 SPARCL1
## 2 ENSG00000109906 ZBTB16
## 3 ENSG00000179094 PER1
## 4 ENSG00000189221 MAOA
## 5 ENSG00000120129 DUSP1
## 6 ENSG00000096060 FKBP5
DEG_edgeR <- cbind(GeneID=rownames(DEG_edgeR),DEG_edgeR)
DEG_edgeR_symbol <- merge(id2symbol,DEG_edgeR,
by.x="ENSEMBL",by.y="GeneID",all.y=T)
head(DEG_edgeR_symbol)
## ENSEMBL SYMBOL logFC logCPM LR PValue FDR
## 1 ENSG00000000003 TSPAN6 -0.38435667 5.055490 4.08601716 0.04323942 0.2651214
## 2 ENSG00000000419 DPM1 0.19874192 4.610192 1.38492762 0.23926357 0.6546538
## 3 ENSG00000000457 SCYL3 0.02784654 3.482541 0.01840403 0.89208891 0.9777599
## 4 ENSG00000000460 FIRRM -0.12127561 1.481403 0.12072575 0.72824868 0.9392750
## 5 ENSG00000000971 CFH 0.43597022 8.088237 3.20730408 0.07331018 0.3638136
## 6 ENSG00000001036 FUCA2 -0.24682279 5.908012 2.37468922 0.12331614 0.4770370
## regulated
## 1 normal
## 2 normal
## 3 normal
## 4 normal
## 5 normal
## 6 normal
# 方法2:gtf文件中得到的id与name关系
# Assembly: GRCh37(hg19) Release: ?
# 使用上课测试得到的count做
# 选择显著差异表达的结果
library(tidyverse)
DEG_edgeR_symbol_Sig <- filter(DEG_edgeR_symbol,regulated!="normal")
# 保存
write.csv(DEG_edgeR_symbol,"result/4.DEG_edgeR_all.csv", row.names = F)
write.csv(DEG_edgeR_symbol_Sig,"result/4.DEG_edgeR_Sig.csv", row.names = F)
save(DEG_edgeR_symbol,file = "data/Step03-edgeR_nrDEG.Rdata")
##====== 检查是否上下调设置错了
# 挑选一个差异表达基因
head(DEG_edgeR_symbol_Sig)
## ENSEMBL SYMBOL logFC logCPM LR PValue
## 1 ENSG00000001626 CFTR -1.0294143 -1.4417818 4.305790 3.798286e-02
## 2 ENSG00000003096 KLHL13 -0.9240690 4.1526529 18.481210 1.715877e-05
## 3 ENSG00000003137 CYP26B1 -1.1464055 2.3133715 5.377759 2.039506e-02
## 4 ENSG00000003402 CFLAR 1.1780649 6.8960435 44.323746 2.783196e-11
## 5 ENSG00000003987 MTMR7 0.9810876 0.3462333 6.599148 1.020276e-02
## 6 ENSG00000004799 PDK4 2.5421303 5.4173139 16.467822 4.948281e-05
## FDR regulated
## 1 2.443426e-01 down
## 2 5.888760e-04 down
## 3 1.635214e-01 down
## 4 4.359678e-09 up
## 5 1.011711e-01 up
## 6 1.443029e-03 up
exp <- c(t(express_cpm[match("ENSG00000001626",rownames(express_cpm)),]))
test <- data.frame(value=exp, group=group_list)
ggplot(data=test,aes(x=group,y=value,fill=group)) + geom_boxplot()
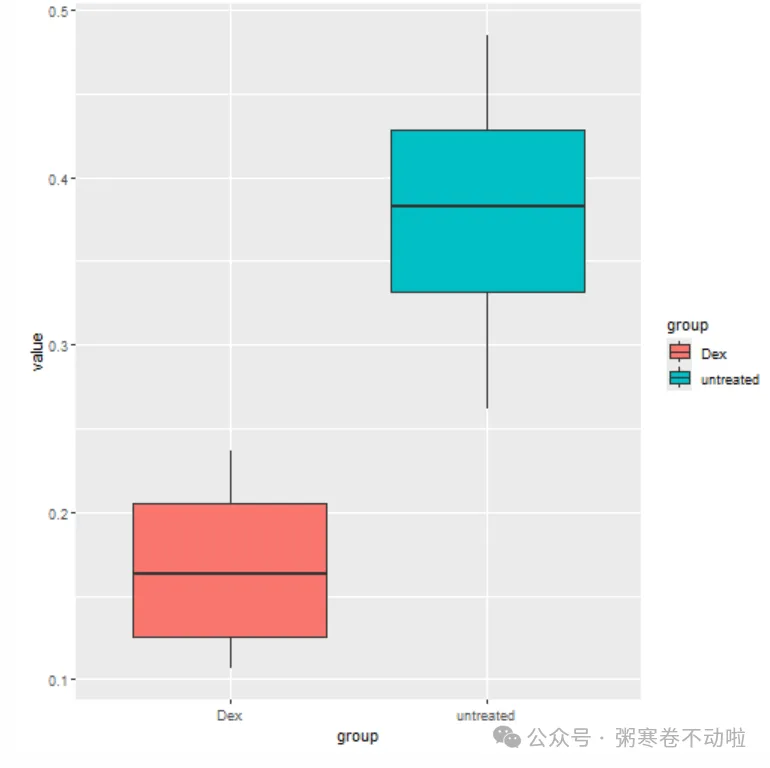
如果想在服务器上使用RStudio怎么操作?
- 如果服务器上安装了RStudio,可以用在线的RStudio Server,和本地一样使用
- 传参脚本
Rscript edgeR.R --count filter_count.txt --group group.txt --comp Dex_vs_untreated --fc 1.5 -- pvalue 0.05 --od ./
nohup sh DEG.sh >DEG.log &
差异分析热图
rm(list = ls())
options(stringsAsFactors = F)
# 加载包
library(pheatmap)
library(tidyverse)
# 加载原始表达矩阵
lname <- load(file = "data/Step01-airwayData.Rdata")
lname
## [1] "filter_count" "express_cpm" "group"
express_cpm1 <- rownames_to_column(as.data.frame(express_cpm) ,var = "ID")
# 读取差异分析结果
lname <- load(file = "data/Step03-edgeR_nrDEG.Rdata")
lname
## [1] "DEG_edgeR_symbol"
# 提取所有差异表达的基因名
edgeR_sigGene <- DEG_edgeR_symbol[DEG_edgeR_symbol$regulated!="normal",]
head(edgeR_sigGene)
## ENSEMBL SYMBOL logFC logCPM LR PValue
## 14 ENSG00000001626 CFTR -1.0294143 -1.4417818 4.305790 3.798286e-02
## 29 ENSG00000003096 KLHL13 -0.9240690 4.1526529 18.481210 1.715877e-05
## 30 ENSG00000003137 CYP26B1 -1.1464055 2.3133715 5.377759 2.039506e-02
## 34 ENSG00000003402 CFLAR 1.1780649 6.8960435 44.323746 2.783196e-11
## 38 ENSG00000003987 MTMR7 0.9810876 0.3462333 6.599148 1.020276e-02
## 55 ENSG00000004799 PDK4 2.5421303 5.4173139 16.467822 4.948281e-05
## FDR regulated
## 14 2.443426e-01 down
## 29 5.888760e-04 down
## 30 1.635214e-01 down
## 34 4.359678e-09 up
## 38 1.011711e-01 up
## 55 1.443029e-03 up
data <- merge(edgeR_sigGene,express_cpm1,by.x = "ENSEMBL",by.y = "ID")
data <- na.omit(data)
data <- data[!duplicated(data$SYMBOL),]
# 绘制热图
dat <- select(data,starts_with("SRR"))
rownames(dat) <- data$SYMBOL
dat[1:4,1:4]
## SRR1039508 SRR1039509 SRR1039512 SRR1039513
## CFTR 0.4846623 0.1063594 0.3551509 0.1319257
## KLHL13 26.6564280 16.7515999 21.9798962 11.2796506
## CYP26B1 6.0098129 3.0312419 10.1020708 4.1556608
## CFLAR 69.0159154 139.1712287 68.7414347 155.9362229
anno <- data.frame(group=group$sample_title,row.names = group$run_accession)
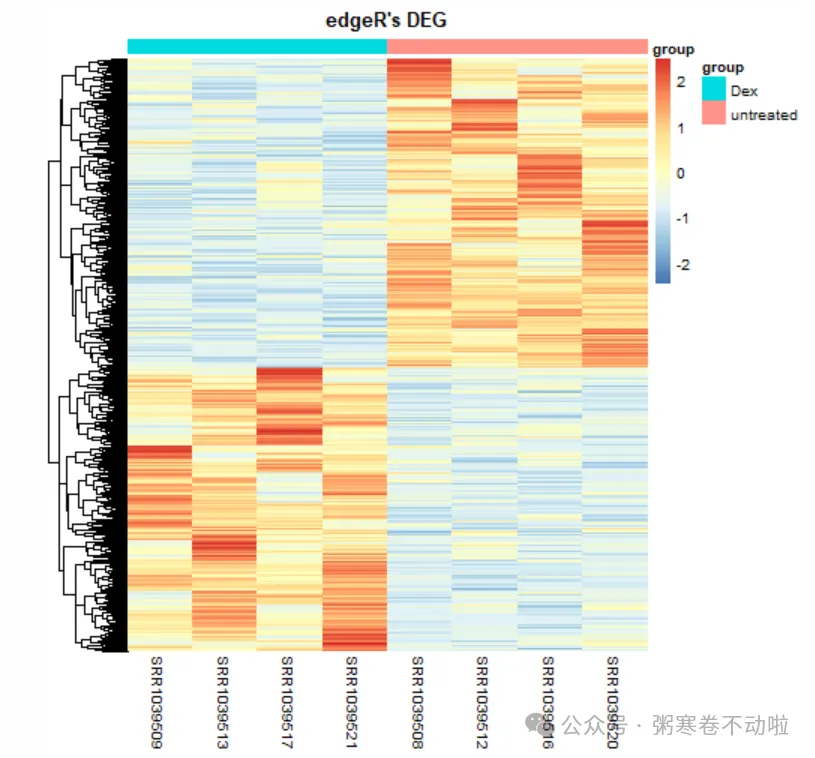
pheatmap(dat,scale = "row",show_colnames =T,
show_rownames = F, cluster_cols = T,
annotation_col=anno,
main = "edgeR's DEG")
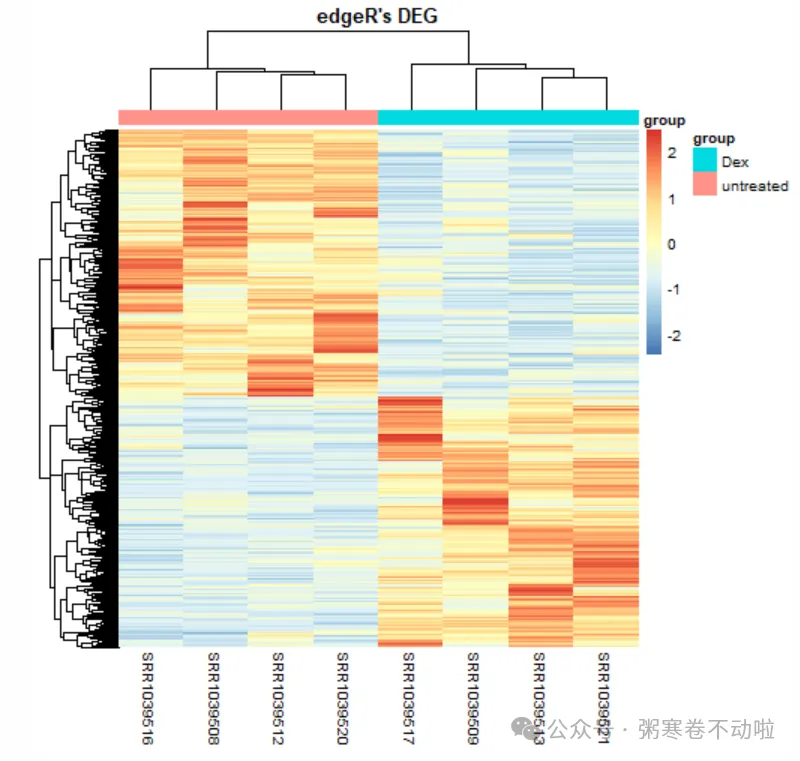
# 显示指定symbol,这里随便展示10个基因symbol
labels <- rep(x = "",times=nrow(dat))
labels[1:10] <- rownames(dat)[1:10]
pheatmap(dat,scale = "row",show_colnames =T,
show_rownames = T,
cluster_cols = T,
annotation_col=anno,
labels_row = labels,
fontsize_row = 8,
main = "edgeR's DEG")
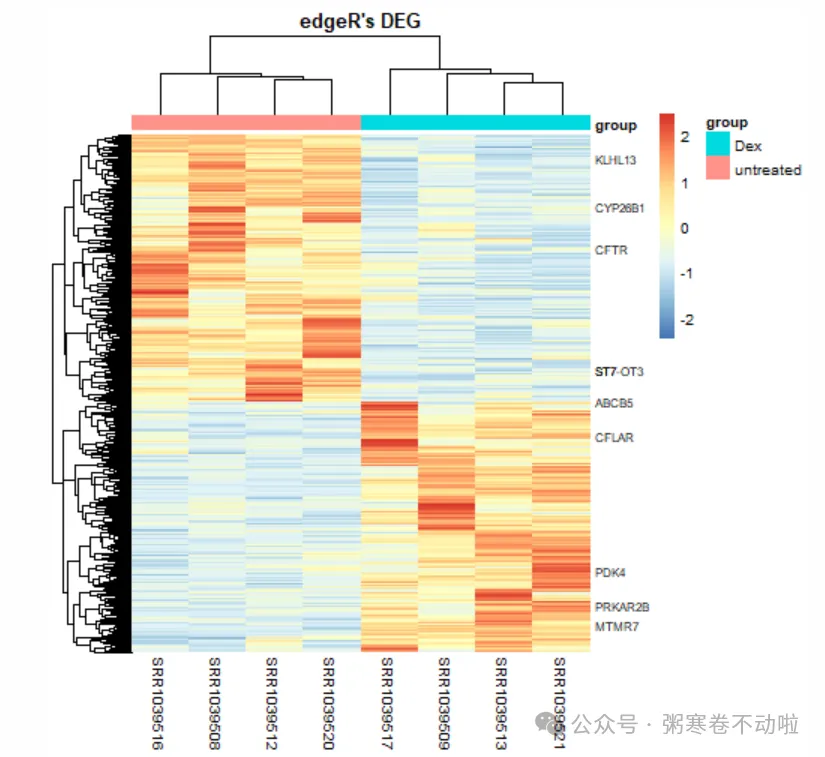
# 按照指定顺序绘制热图
dex_exp <- express_cpm[,match(rownames(anno)[which(anno$group=="Dex")],
colnames(express_cpm))]
untreated_exp <- express_cpm[,match(rownames(anno)[which(anno$group=="untreated")],
colnames(express_cpm))]
data_new <- cbind(dex_exp, untreated_exp)
dat1 <- data_new[match(edgeR_sigGene$ENSEMBL,rownames(data_new)),]
pheatmap(dat1, scale = "row",show_colnames =T,show_rownames = F,
cluster_cols = F,
annotation_col=anno,
main = "edgeR's DEG")
差异分析火山图
rm(list = ls())
options(stringsAsFactors = F)
library(ggplot2)
library(tidyverse)
# 读差异分析结果
lname <- load(file = "data//Step03-edgeR_nrDEG.Rdata")
# 根据需要修改DEG的值
data <- DEG_edgeR_symbol
colnames(data)
## [1] "ENSEMBL" "SYMBOL" "logFC" "logCPM" "LR" "PValue"
## [7] "FDR" "regulated"
# 绘制火山图
colnames(data)
## [1] "ENSEMBL" "SYMBOL" "logFC" "logCPM" "LR" "PValue"
## [7] "FDR" "regulated"
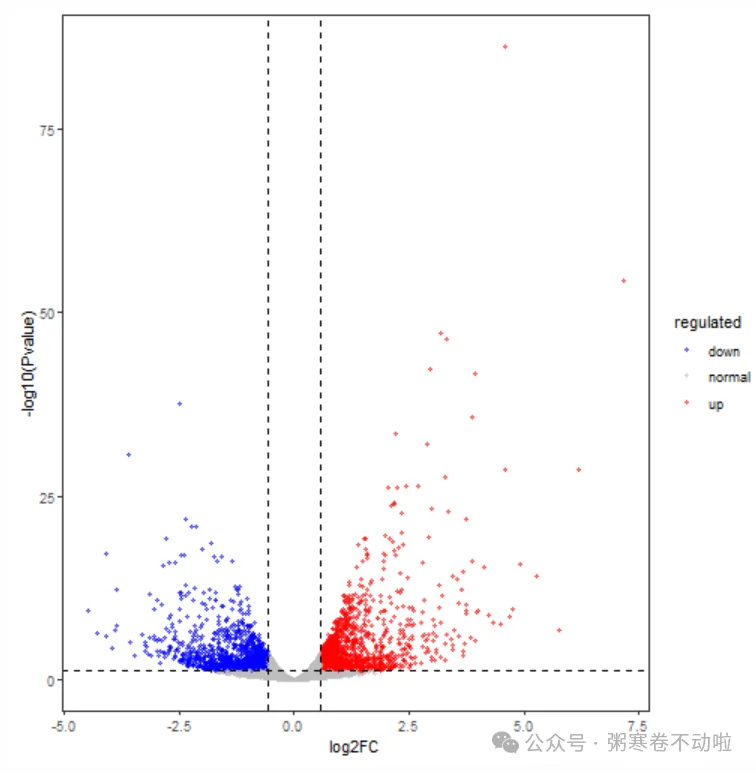
p <- ggplot(data=data, aes(x=logFC, y=-log10(PValue),color=regulated)) +
geom_point(alpha=0.5, size=1.2) +
theme_set(theme_set(theme_bw(base_size=20))) + theme_bw() +
theme(panel.grid.major=element_blank(),panel.grid.minor=element_blank()) +
xlab("log2FC") + ylab("-log10(Pvalue)") +
scale_colour_manual(values = c(down='blue',normal='grey',up='red')) +
geom_vline(xintercept=c(-(log2(1.5)),log2(1.5)),lty=2,col="black",lwd=0.6) +
geom_hline(yintercept = -log10(0.05),lty=2,col="black",lwd=0.6)
p
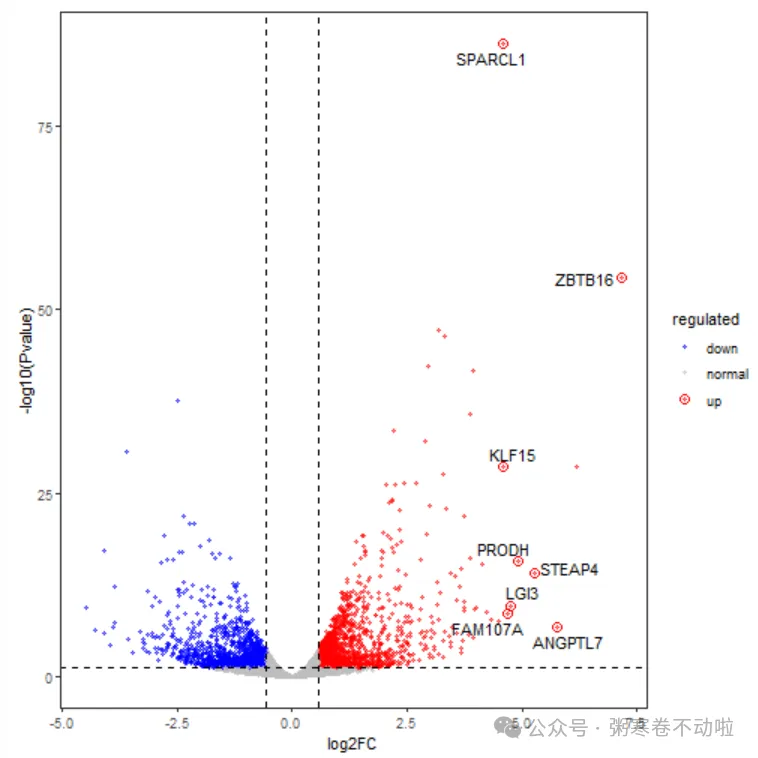
# 添加top基因
# 通过FC选取TOP10
label <- data[order(abs(data$logFC),decreasing = T)[1:10],]
# 通过pvalue选取TOP10
#label <- data[order(abs(data$PValue),decreasing = F)[1:10],]
label <- na.omit(label)
p1 <- p + geom_point(size = 3, shape = 1, data = label) +
ggrepel::geom_text_repel( aes(label = SYMBOL), data = label, color="black" )
p1
# 保存结果
png(file = "result/5.Volcano_Plot.png",width = 900, height = 800, res=150)
plot(p1)
dev.off()
## png
## 2
from 生信技能树
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-06-24,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号