单细胞数据分析新选择(基于Julia编程语言)

单细胞数据分析新选择(基于Julia编程语言)

作为数据分析语言三剑客之一的Julia,一直以来在生物学领域并没有得到太多的重视。但在数学、物理、化学以及工程计算领域,Julia语言的应用范围一直在不断拓展。2023年Elisabeth Roesch等人在文章Julia for Biologists中为生物学家使用Julia的方方面做了一个较为详尽的概述,从一个宏观的角度推动生物计算开发者尝试这门拥有多种特性的编程语言。
ASCT: Automatic Single-Cell Toolbox
基于对Julia语言的开发兴趣,以及想要建立一个尽量为用户提供自动化参数选择,同时保持灵活性的单细胞数据下游分析工具,西湖大学高性能计算中心开源了一个100%由Julia编写的工具ASCT。该工具目前在github上发布了完全开源的0.9.0版本,并提供了基本分析流程和使用Harmony算法实现的数据整合流程。该工具基本对标R上的Seurat v4版本,并能对不同来源的数据进行一些自适应的参数设置而无需用户自行摸索。本文以其提供的pbmc3k流程来大致介绍该工具的基本使用方法。
ASCT安装
在Julia中进入包管理界面后,可以通过网络直接安装github上ASCT包(所以需要大家自行掌握基本的Julia编程基础哦,包括安装Julia,以及使用Julia的IDE)
add https://github.com/kaji331/ASCT
安装好后退出包管理界面,使用下面的命令就可以顺利载入ASCT包
using AutomaticSingleCellToolbox
读取Cellranger结果
在ASCT中,目前暂时只支持使用最多的Cellranger结果的读取,同时一个函数既可以读取传统的matrix.mtx目录,也可以读取单一的h5格式文件。
pbmc = Read10X("filtered_gene_bc_matrices/hg19";min_cells=3)
这条命令在读取hg19下pbmc3k数据的三个文件的同时,过滤表达细胞数量小于3个的基因。单细胞表达数据以及相关的信息都被保存在名为pbmc的数据结构中,该数据结构是ASCT针对单细胞RNA数据建立的名为WsObj的mutable struct。
https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz (复制粘贴这个网页地址去浏览器打开即可下载哦)
数据质控
通过FeaturePercentage!函数,ASCT直接修改WsObj结构,添加特定基因集在每个细胞中的表达比例结果。该函数默认通过正则表达式选定人或小鼠的线粒体基因集,也可以直接输入一个包含特定基因集名称的向量Vector来进行计算。生成的结果默认保存变量名为percentage_mt。
FeaturePercentage!(pbmc)
接下来用户可以使用DrawQC这个函数来绘制单细胞数据的质控图(对于ASCT的目标来说,其实用户已经没有绘制质控图的必要了)。
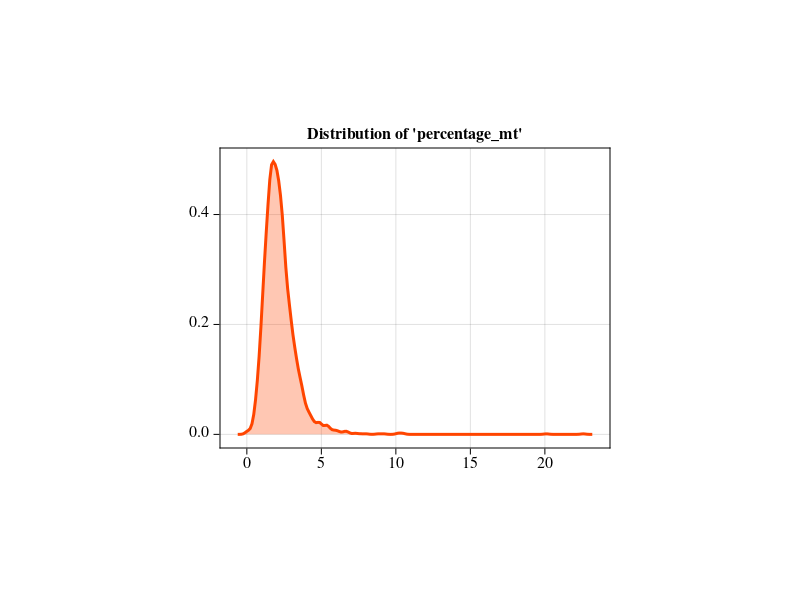
DrawQC(pbmc;obs_name="percentage_mt")
DrawQC(pbmc)
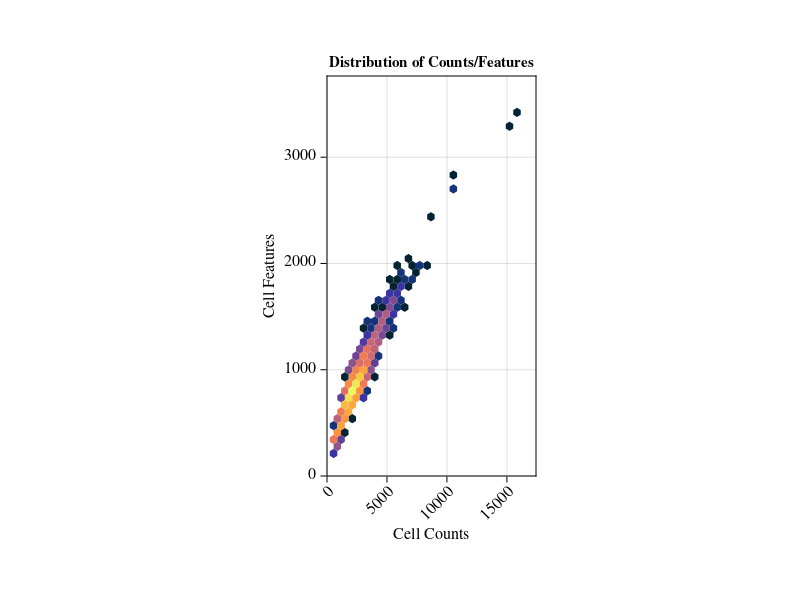
通过这两个质控图,我们可以看到pbmc这个数据中线粒体基因在不同细胞中表达比例的分布,以及细胞中UMI数量和基因的相对分布情况。用户可以使用ManualFilter!函数像Seurat或者Scanpy一样去看图设定过滤参数并进行细胞的过滤,但ASCT更为推荐用户使用
AutoFilter!(pbmc)
来进行自动过滤。该函数默认对线粒体基因和UMI/Featue进行过滤。
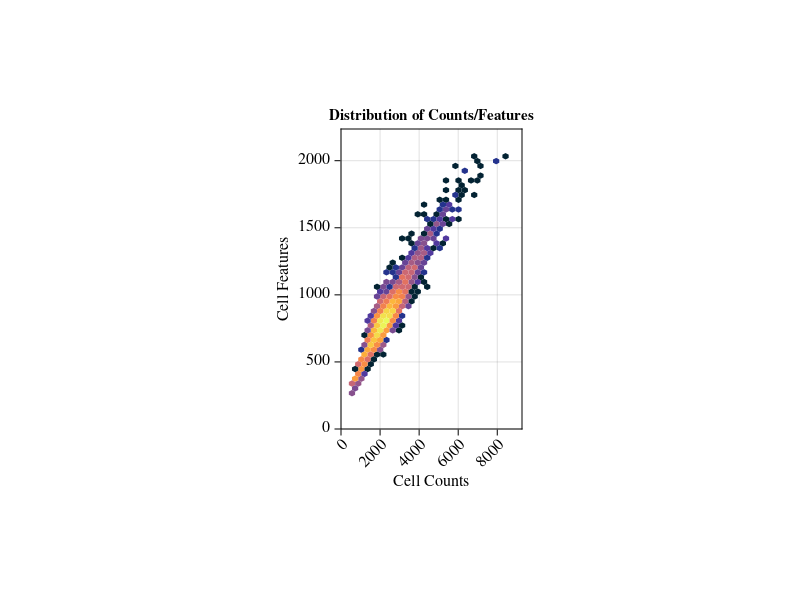
从图中可以看到ASCT自动寻找参数并完成了过滤,我们认为效果是优于用户手动过滤的,尤其是单细胞分析入门用户如果盲目的设定过滤参数可能导致的重要细胞丢失或干扰细胞的引入。
降维聚类
接下来ASCT使用NormalizeData!和SelectHVG!来进行数据的标准化和高变基因的选取,这两个步骤一般也不需要用户进行参数的调整。ASCT的PCA!函数会对数据先进行归一化,再计算PCA,或者直接利用FastRowScale!已经算好的归一化数据。
PCA!(pbmc)
UMAP!(pbmc)
TSNE!(pbmc;fast=false)
UMAP和t-SNE的计算都属于可选步骤,用户可以自行选择是否都执行。其中TSNE!默认使用FIt-SNE来进行计算(即fast=true),不过用户需要提前安装FIt-SNE应用程序。
通过
ElbowPCA(pbmc)
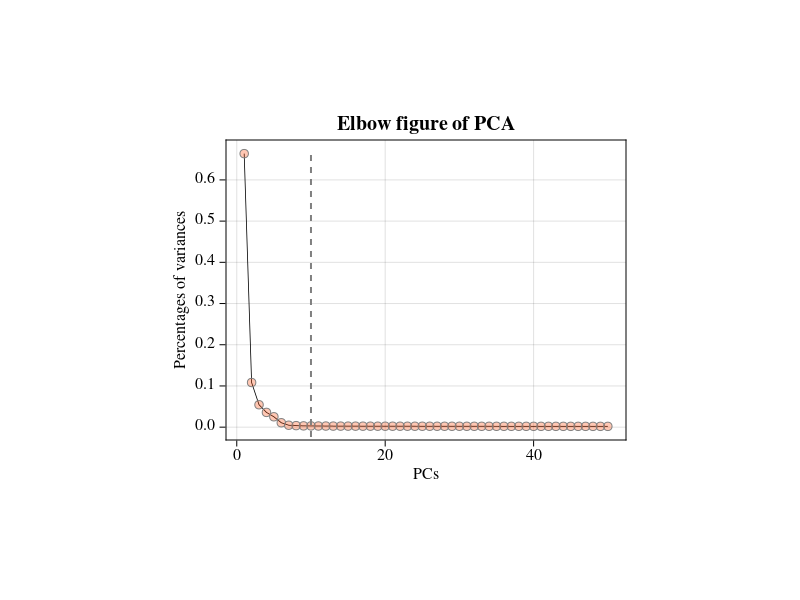
我们可以看到PCA!函数在执行过程中已经自动选择了合理的PC数量并自动应用到了后续的UMAP!和TSNE!计算中。
聚类
ASCT提供了Louvain和k-medoids两种算法来进行聚类,默认使用Louvain算法,并且基于R包clustree的思想来自动推定resolution。
Clustering!(pbmc)
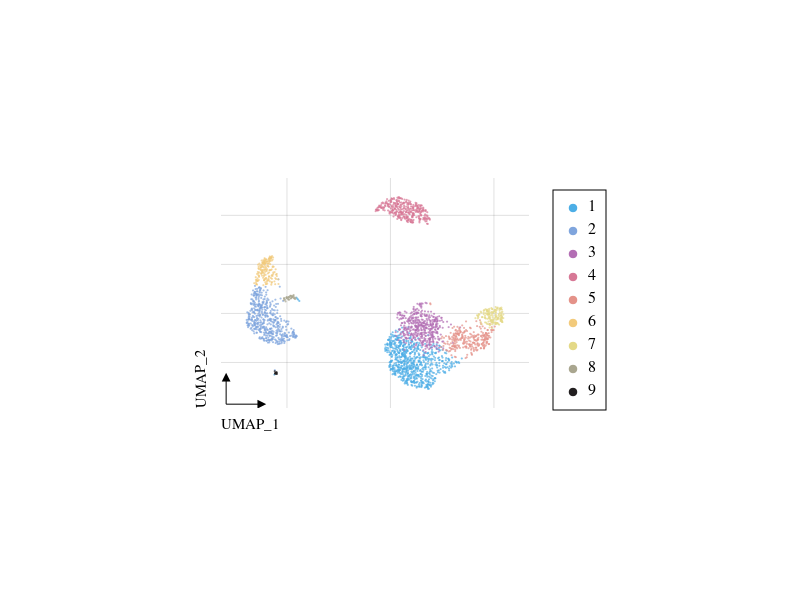
DimensionPoints(pbmc;dimension_name="umap")
[ Info: Max modularity: 0.87166995
[ Info: Recommended resolution is 0.4
Marker基因的选择
ASCT用Julia实现了COSG算法并默认采用该算法来寻找所有cluster的marker基因,特异性好,速度快。同时支持Wilcox检验、t检验和基于广义线性模型的似然率检验。(所以并不是R编程语言那样需要去专门安装github里面的COSG算法的对应的包啦 )
@time DE!(pbmc)
2.104334 seconds (1.09 M allocations: 1.291 GiB, 4.96% gc time)
"Finished!"
使用Julia重新实现的COSG算法速度不输于原版C/C++的实现,在同一台测试电脑上两者都能在1-2秒内完成pbmc数据的marker基因搜寻。
?DE!
查看DE!函数的帮助信息,我们可以知道ASCT的DE!可以通过trt/ctl参数来设定不同分组之间进行比较,同时使用group_name来设置分组变量的名称,还能通过sub_group参数来限定只对某个特定批次的样本搜寻marker基因等多样化的分析。
using DataFrames
groupby(pbmc.meta["clusters_latest_DE"],:group) |>
x -> combine(x) do y
first(y,10)
end
marker基因的结果保存在pbmc的meta成员中,类型为DataFrame,因此可以使用DataFrames包的combine操作做进一步的查看和处理。
group | gene | score | pct1 | log2fc |
|---|---|---|---|---|
String | String | Float64 | Float64 | Float64 |
1 | CCR7 | 0.41635 | 0.423181 | 1.3035 |
1 | LDHB | 0.319986 | 0.893531 | 1.07722 |
1 | LDLRAP1 | 0.24444 | 0.234501 | 1.05686 |
... | ... | ... | ... | ... |
可视化
除了上面已经展示的方法外,ASCT目前已经支持的可视化方法还包括:
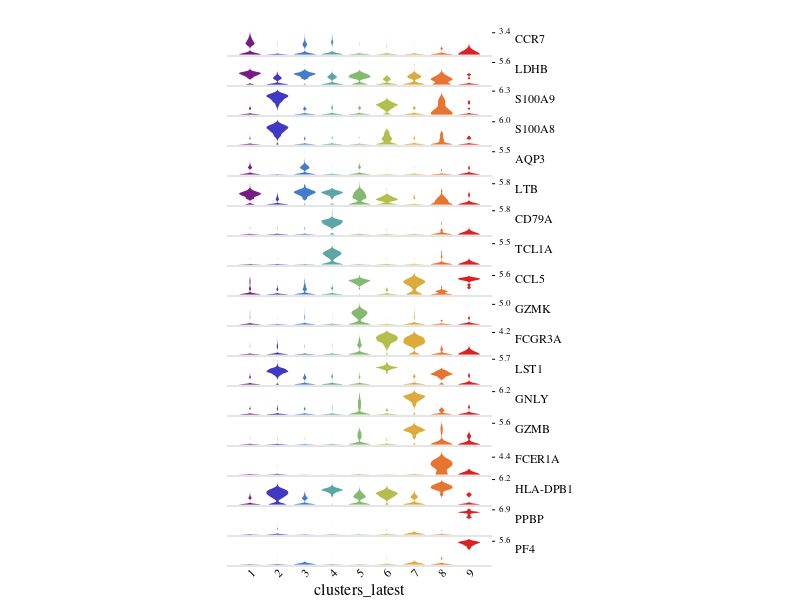
对DE!结果的小提琴图
FeatureViolin(pbmc)
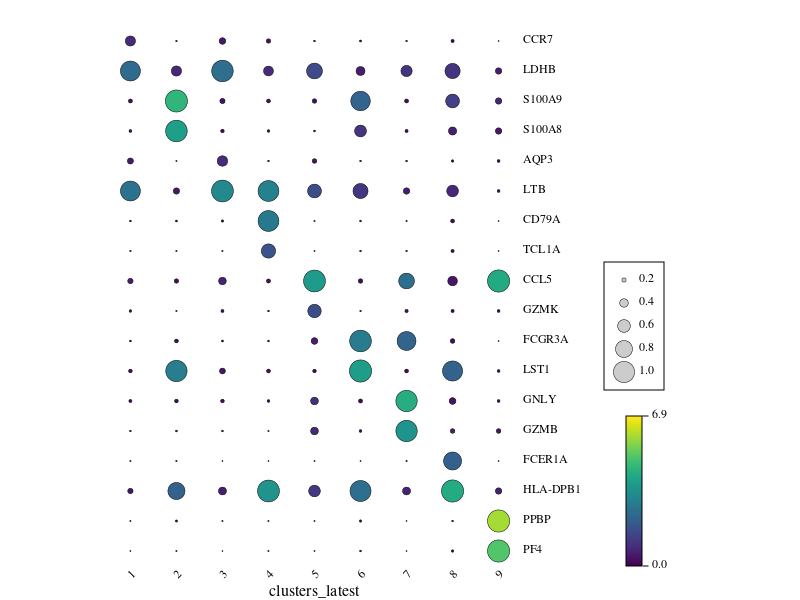
气泡图
FeatureFracDots(pbmc)
热图
FeatureHeat(pbmc;gene_label_size=3)
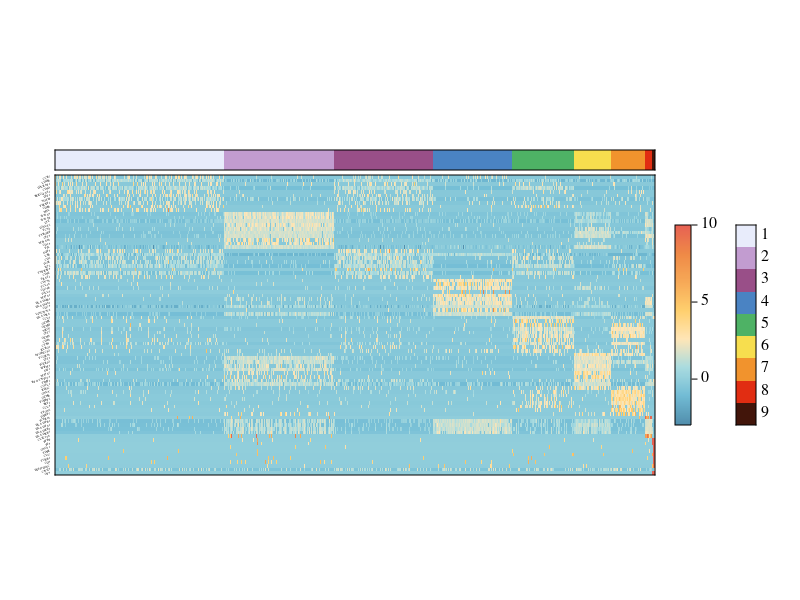
ASCT的可视化基于CairoMakie体系,具有丰富的灵活性。高级用户可以尝试对生成的图片进行个性化的调整。如果是R编程语言,其实Seurat输出的图表是ggplot2体系,所以呢很容易个性化自定义,关于Seurat输出的图表以前我们做了一个投票:可视化单细胞亚群的标记基因的5个方法,下面的5个基础函数相信大家都是已经烂熟于心了:
- VlnPlot(pbmc, features = c("MS4A1", "CD79A"))
- FeaturePlot(pbmc, features = c("MS4A1", "CD79A"))
- RidgePlot(pbmc, features = c("MS4A1", "CD79A"), ncol = 1)
- DotPlot(pbmc, features = unique(features)) + RotatedAxis()
- DoHeatmap(subset(pbmc, downsample = 100), features = features, size = 3)
对比起来, 另外一个Python编程语言的scanpy的可视化也做的很棒。
数据交换
在Julia中,推荐用户使用JLD2来将计算过程的变量保存到基于HDF5的JLD2文件中,可以实现快速的保存和读取,以方便随时恢复当前的分析进度。ASCT也为熟悉Seurat和Scanpy的用户提供了
SaveSeuratV4(pbmc,"pbmc3k.h5seurat")
SaveAnnData(pbmc,"pbmc3k.h5ad")
两种方法将现有的分析结果导入到R语言或Python语言环境中,方便利用已有的其他下游分析工具进行个性化分析。
文档
ASCT的文档目前还比较简陋,github上主要以三个示例ipynb文件为主,西湖大学高性能计算中心将不断的进行丰富。用户目前可以通过在Julia中输入AutomaticSingleCellToolbox.并按Tab键来查看所有的包成员,例如
AutomaticSingleCellToolbox.WsObj |> dump
AutomaticSingleCellToolbox.WsObj <: Any
dat::Dict{AbstractString, AbstractMatrix}
obs::DataFrames.DataFrame
var::DataFrames.DataFrame
log::Vector{AbstractString}
meta::Union{Nothing, Dict{AbstractString, Union{AbstractString, Real, DataFrames.DataFrame, AbstractMatrix, AbstractVector}}}
可以看到WsObj数据结构的完整形态。还可以使用
?AutomaticSingleCellToolbox.RegressObs!
看到如何使用该函数来消除线粒体基因对表达水平的影响,以及
?AutomaticSingleCellToolbox.FeatureScore!
如何给细胞按基因集进行打分和分类……
腾讯云开发者
