读书笔记 | 第二部分 NGS 介绍和数据分析


Part II Introduction to Next-Generation Sequencing (NGS) and NGS Data Analysis 4 Next-Generation Sequencing (NGS) Technologies: Ins and Outs
4.1 How to Sequence DNA: From First Generation to the Next
4.1 如何测序DNA:从第一代到下一代
para
- DNA分子中核苷酸的序列可以通过多种方式确定。
- 在20世纪70年代初,生物化学家(沃尔特·吉尔伯特博士和弗雷德里克·桑格博士)设计了不同的方法来测序DNA。
- 吉尔伯特博士的方法基于化学程序,能够特异性地在四种碱基的每一个位置上分解DNA。
- 另一方面,桑格博士的方法利用了DNA合成过程。
- 在这个过程中,新的DNA链是逐个碱基合成的,使用模板上的序列信息(第2章)。
- 在桑格博士的方法中,使用化学修饰的核苷酸,即双脱氧核苷酸,作为不可逆的DNA链终止剂,随机地在每个碱基位置停止合成过程,从而产生一系列长度不同、相差一个碱基的新DNA链(图4.1)。
- 通过确定吉尔伯特博士方法中特异性分解的DNA分子的单碱基分辨率长度,或桑格博士方法中在每个四种核苷酸随机终止的新DNA链的长度,使得模板DNA的测序成为可能。
- 多年来,桑格方法得到了进一步发展。
- 将自动化集成到该过程中,减少了人工参与并提高了效率。
- 使用荧光标记的终止剂,而不是最初使用的放射性标记终止剂,使得操作更安全,序列检测更稳健。
- 通过使用毛细管电泳而不是平板凝胶来改进DNA链的分离,使得碱基识别具有高置信度。
- 所有这些发展使得桑格方法被广泛采用,并成为人类基因组项目的首选方法。
- 即使在今天,这种方法仍然广泛用于单次或低通量DNA测序。
- 随着下一代测序(NGS)的出现,这种方法已成为第一代测序的同义词。
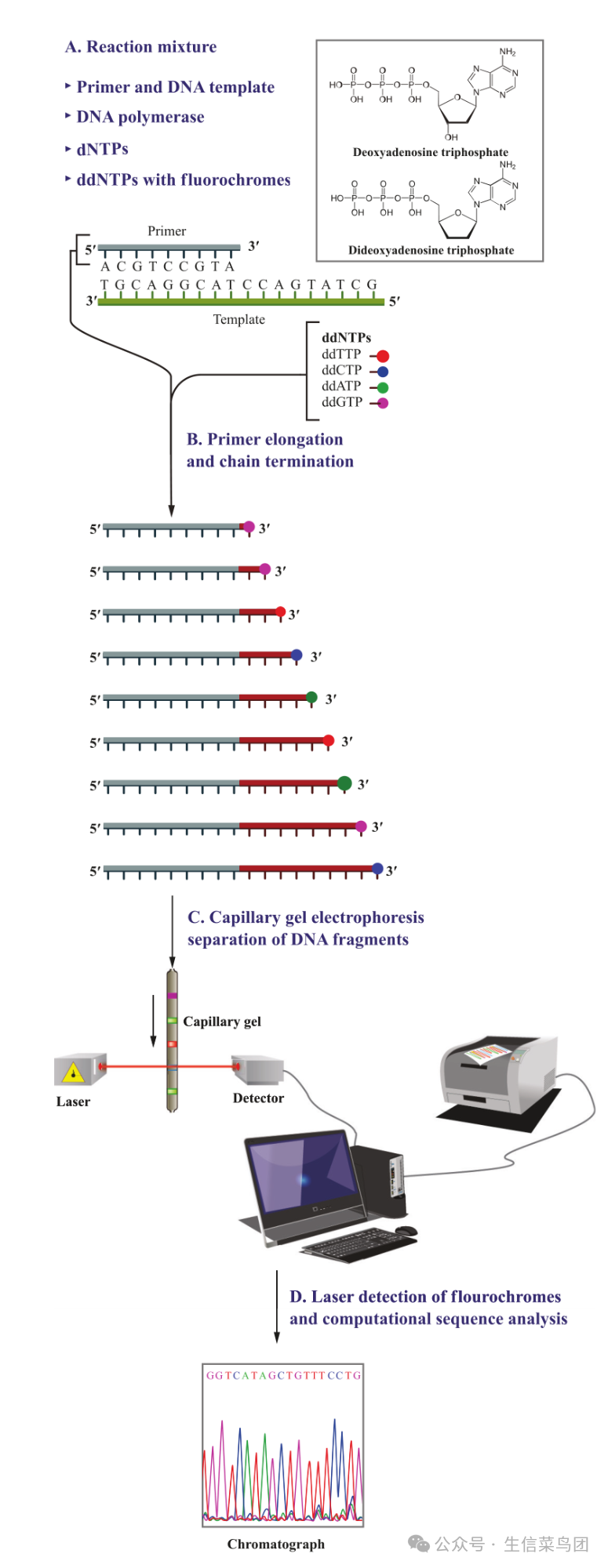
image-20241028010615389
- 图4.1 原始提议的桑格测序方法。该方法包括一个新DNA链合成的步骤,使用测序目标DNA作为模板,随后通过解析新合成的DNA链进行序列推断。
- 在第一步(A)中,新链合成反应混合物包含变性DNA模板、引物、DNA聚合酶和dNTPs。
- 除了dNTPs,桑格方法的特征在于使用标记有不同荧光染料的双脱氧核苷酸(ddG、ddA、ddT和ddC;插图展示了ddATP和dATP的结构差异)。
- 反应混合物中的DNA聚合酶将双脱氧核苷酸与常规核苷酸一同掺入延长的DNA链中,但一旦掺入双脱氧核苷酸,链的延长即终止。
- 在这种测序方案中,这些双脱氧核苷酸与其常规对应物的比例受到控制,以便聚合作用可以在每个碱基位置随机终止。
- 最终产物是一系列不同长度的DNA片段,每个片段的长度取决于双脱氧核苷酸的掺入位置。
- 这些片段随后通过毛细管电泳分离,其中较小的片段比较大的片段迁移得更快,因此更早通过激光检测器。
- 它们携带的荧光染料标签使计算机能够推断出原始DNA的具体序列。(图片改编自https://commons.wikimedia.org/w/index.php?curid=23264166,由Estevezj提供。在知识共享署名-相同方式共享3.0未ported(CC BY-SA 3.0)许可下使用(https://creativecommons.org/licenses/by-sa/3.0/deed.en)。)
para
- 尽管它在测序单个DNA片段方面非常强大,但Sanger方法难以实现高通量,这是降低测序成本的关键,主要是因为其DNA合成过程与随后的DNA链分离/检测过程的分离。
- 然而,其基于合成测序的原理成为许多下一代测序(NGS)技术的基础,包括Illumina的可逆终止子测序、Pacific Biosciences的单分子实时(SMRT)测序、ThermoFisher的Ion Torrent半导体测序以及已停用的454/Roche的焦磷酸测序。
- 与第一代方法不同,这些技术使用带有可逆终止子或其他可切割化学修饰的核苷酸,或常规未修饰的核苷酸,因此新DNA链的合成不会被永久终止,从而可以在每个碱基结合时或之后进行监测。
para
- 并非所有下一代测序(NGS)技术都基于测序-by-合成原理。
- 例如,牛津纳米孔测序和已停产的Life Technologies的SOLiD测序分别使用纳米孔传感和测序-by-连接。
- 尽管不同NGS技术在原理上有所不同,但它们有一个共同点,使它们区别于第一代测序,那就是它们通过同时测序数百万到数十亿个DNA分子,具有巨大的数据吞吐量。
- 除了接下来要详细介绍的新的测序化学或检测方案的开发智慧外,NGS技术在实现极高吞吐量方面的成功还得益于现代工程和计算技术的成就。
- 微流体和微制造方面的进步使得从微量的测序反应中检测信号成为可能。
- 现代光学和成像技术的发展使得高分辨率、高保真度和高速度地追踪测序反应成为可能。
- 一些NGS平台还依赖于半导体行业数十年的进步,或是更近期但迅速发展的纳米孔技术(例如Ion Torrent和Nanopore平台)。
- 高性能计算使得处理和解构从数百万个这些过程中记录的海量信号成为可能。
para
- 由于不同的下一代测序(NGS)技术采用不同的机制和实施策略,在下一节中将详细介绍截至撰写时(2022年初)一些最广泛采用的NGS平台的具体情况。
- 随着NGS技术的不断发展,新的平台将会出现,而一些当前的技术将变得过时。
- 尽管NGS平台的概述通常很快就会过时,但本书中介绍的关于NGS数据分析的指导原则将保持不变。
4.2 Ins and Outs of Different NGS Platforms
4.2 不同NGS平台的优缺点
4.2.1 Illumina Reversible Terminator Short-Read Sequencing
4.2.1 Illumina 可逆终止子短读测序
4.2.1.1 Sequencing Principle
4.2.1.1 测序原理
para
- Illumina NGS平台迄今为止最受欢迎,并生成了最大量的NGS数据。
- Illumina测序技术的核心是使用带有可逆终止剂的荧光标记核苷酸。
- 如前所述,这种方法基于与Sanger方法相同的测序合成基本原理;但与Sanger方法不同的是,在每一个这些特殊修饰核苷酸结合后,它们携带的终止剂部分只是暂时阻止新DNA链的延伸。
- 在基于其特定荧光标记的光学检测结合的核苷酸后,终止剂部分被切割,从而新链合成恢复,进入下一个核苷酸结合循环。
- 为了同时检测数百万到数十亿个测序反应中的核苷酸结合,dATP、dCTP、dGTP和dTTP被标记上不同的荧光标签,以便每个核苷酸可以通过它们发出的不同荧光信号被检测。
- 荧光标签和可逆终止剂部分通过相同的化学键连接到核苷酸上,因此在每个核苷酸结合和检测循环后,它们都可以在单一反应中被切割掉,为下一个核苷酸的结合做准备。
4.2.1.2 Implementation
4.2.1.2 实施
para
- Illumina NGS系统中的测序反应发生在流动池中(图4.2)。
- 流动池中的流体通道,通常称为泳道,是测序反应发生的地方,测序信号通过扫描收集。
- 每个泳道的顶部和底部表面覆盖着一层寡核苷酸序列,这些序列与Illumina适配器中的锚定序列互补。
- 当通过片段化和适配器连接制备的DNA测序文库被加载到每个泳道中时,文库中的DNA模板结合到这些寡核苷酸序列上,并固定在泳道表面(图4.3)。
- 固定后,每个模板分子通过一种称为"桥式扩增"的等温过程进行克隆扩增,通过这一过程,在紧密相邻的区域(直径小于1微米)生成多达1,000个相同的模板拷贝,形成一个簇。
- 在测序过程中,这些簇是基本的检测单元,能够产生足够的信号强度用于碱基识别。

image-20241028010813399
- 图4.2 Illumina测序流动池。它是一个特殊的玻璃载片,内部包含流体通道(称为泳道)。
- 测序文库在模板固定和簇生成后,被加载到泳道中进行大规模并行测序。
- 在测序过程的每一步中,包括DNA聚合酶和修饰过的dNTPs的DNA合成混合物,通过位于两端的入口和出口端口被泵入和泵出每个泳道。
para
- 在理想条件下,同时将核苷酸整合到单个簇中许多相同的测序模板副本中,预期每一步都会同步进行,因此保持相位一致。
- 实际上,少量模板会与同一簇中的大多数分子失去同步,导致要么落后(称为相位偏移),由于终止子未完全去除以及错过一个循环,要么提前一个或几个碱基(预相位偏移),由于整合了没有终止子的核苷酸。
- 簇中存在相位偏移和预相位偏移会导致背景噪声增加和碱基识别质量下降。
- 当进行越来越多的测序循环时,这个问题会变得更糟。
- 这就是为什么基于克隆扩增的平台(也包括稍后详述的Ion Torrent平台)在测序末端的碱基识别质量评分会下降。
- 最终,碱基识别质量的下降会达到一个阈值,超过这个阈值,质量评分变得完全不可接受。
- 同步性的逐渐丧失是这些平台读长的主要决定因素。
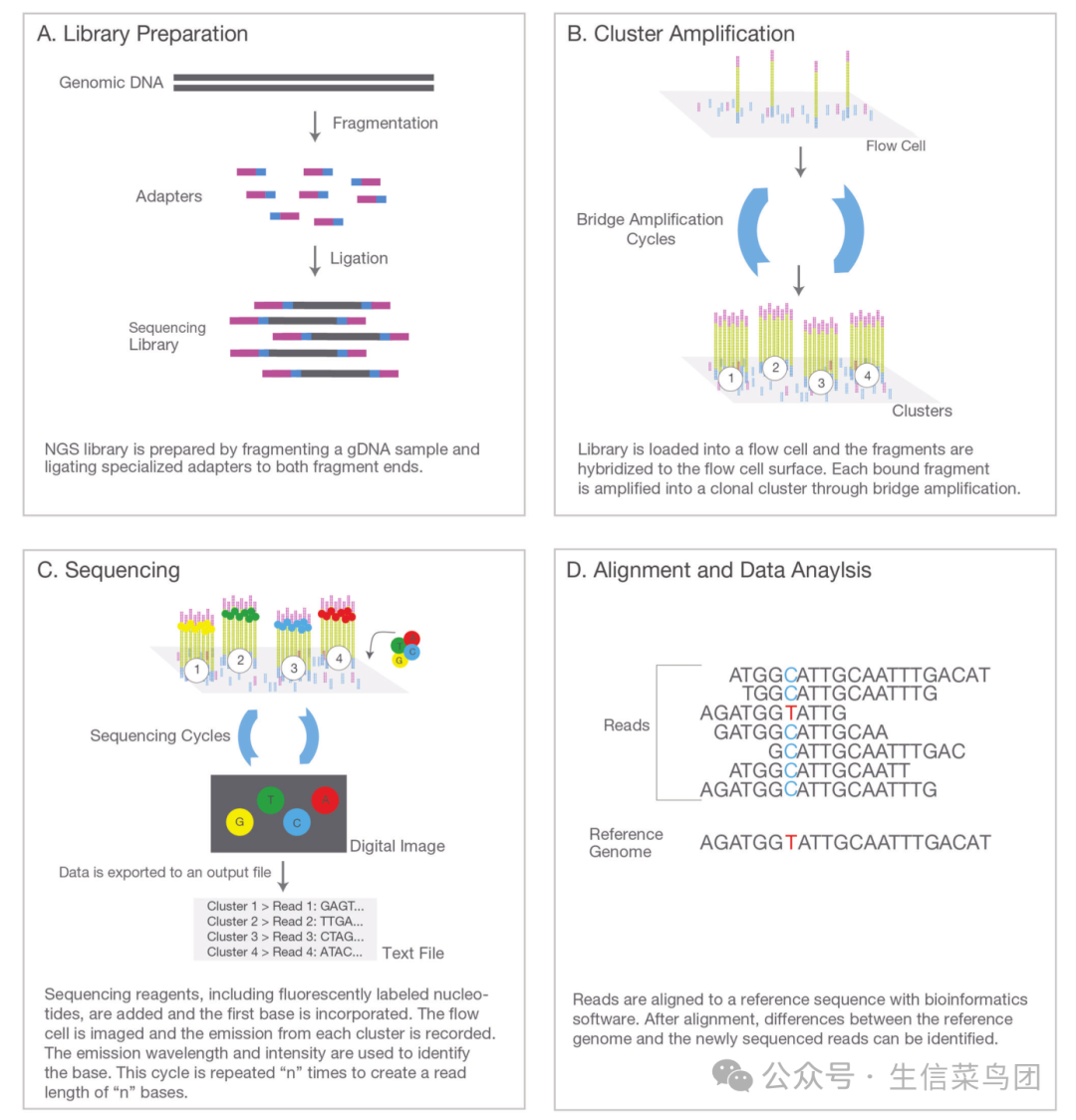
image-20241028010943965
- 图4.3 Illumina测序过程概览。(根据Illumina, Inc.的许可使用。保留所有权利。)
4.2.1.3 Error Rate, Read Length, Data Output, and Cost
4.2.1.3 错误率、读取长度、数据输出和成本
para
- Illumina测序方法的总体错误率低于1%,这使得它成为目前最准确的NGS平台之一。
- 最常见的错误类型是单核苷酸替换。
- 在读取长度方面,截至撰写本文时所有可用的Illumina测序仪都能产生至少150个碱基的读取。
- 一些测序仪可以生成长达250个碱基(NovaSeq 6000)或300个碱基(MiSeq)的读取。
- 除了从DNA模板的一端读取(即单端测序)外,Illumina测序仪还可以从DNA片段的两端读取(称为双端测序)。
- 双端测序不仅使序列读取的总数翻倍,还具有便于后续对参考基因组(详情见下一章)或基因组组装(第12章)进行对齐的优势,从而减少由相对较短的读取长度引起的限制。
para
- 关于数据输出,Illumina 提供的不同测序仪/流动池具有不同的输出水平(见表4.1),其中在NovaSeq 6000上使用S4流动池可以达到最大3 Tb(太碱基)的输出。
- 测序运行时间取决于读长,通常在不到一天到几天不等。
- 在测序成本方面,根据测序仪和流动池类型的不同,生产规模测序仪(即NovaSeq 6000和NextSeq 2000)上每Gb数据的成本为4.80至41.67美元。
- 这一成本计算基于当前测序试剂的标价除以相应的数据输出,因此不包括其他成本,如文库制备试剂、人员时间、测序仪折旧和服务合同等。
- 还应注意的是,此处提供的数字截至2022年初,并将随着未来系统更新而变化。
4.2.1.4 Sequence Data Generation
4.2.1.4 序列数据生成
para
- Illumina序列数据生成过程有三个步骤。
- 首先,每个循环后捕获的原始图像被分析,以定位簇并报告每个簇的信号强度、坐标和噪声水平。
- 这一步骤由仪器控制软件执行。
- 这一步骤的输出被输入到下一个碱基识别步骤,由仪器的实时分析(RTA)软件执行,该软件使用簇的信号强度和噪声水平进行碱基识别和质量分数计算。
- 这一步骤还会过滤掉低质量的读取。
- 在第三步骤中,碱基识别文件,即bcl文件,被转换为FASTQ文件,其中包含原始读取。
- 由于通常多个样本以多重方式一起测序,因此在第三步骤中还进行序列数据的解复用。
- 这通常使用Illumina的bcl2fastq工具执行,但也可以使用其他工具,如IlluminaBasecallsToFastq。
- 解复用后的FASTQ文件以压缩格式存储,这是最终用户在NGS设施完成运行后通常收到的文件。
4.2.2 Pacific Biosciences Single-Molecule Real-Time (SMRT) Long-Read Sequencing
4.2.2 太平洋生物科学单分子实时(SMRT)长读测序
4.2.2.1 Sequencing Principle
4.2.2.1 序列化原则
para
- 太平洋生物科学公司的SMRT测序平台通常被认为是第三代测序技术,因为它足够灵敏,能够测序单个DNA分子,因此可以绕过扩增过程。
- 此外,该平台生成的读长比Illumina NGS平台要长得多,目前的读长可达25 kb及以上。
- 虽然它也是基于测序合成原理,但与Illumina方法不同,SMRT测序使用携带独特荧光标签的核苷酸,这些标签连接在其末端磷酸基团上,但没有终止基团。
- 当一个核苷酸被整合到延长的DNA链中时,末端磷酸基团(实际上是前面提到的焦磷酸基团)的裂解会同时释放荧光标签,这使得能够进行实时信号检测。
- 由于这个过程不涉及单独的荧光标签释放和检测步骤,测序检测信号是连续记录为一部电影,最长可达30小时,而不是使用扫描图像。
4.2.2.2 Implementation
4.2.2.2 实施
para
- PacBio测序的核心是SMRT细胞,它携带数百万个孔,技术上称为零模波导(或ZMWs),用于同时测序数百万个DNA模板(截至2022年初的当前版本有800万个ZMWs)。
- ZMWs本质上是在100纳米厚的金属薄膜中微制造的直径为几十纳米的孔,该金属薄膜又沉积在玻璃基板上。
- 由于ZMW的直径小于可见光的波长,并且可见光通过如此小的开口从玻璃底部自然通过,只有ZMW底部的30纳米被照亮。
- 拥有仅为20泽普升(10⁻²¹升)的检测体积,这种检测方案大大减少了背景噪声,并能够检测到不同波长的光,这些光是由核苷酸掺入新DNA链时发出的。
para
- 尽管SMRT平台进行单分子测序,但标准的文库制备协议仍然需要以µg水平的DNA样本作为起始(对于较低的DNA输入需要进行扩增)。
- 文库制备过程包括将DNA片段化至所需长度、末端修复/A尾添加,以及连接发夹环适配器。
- 这导致了被称为SMRTbell的环形结构的形成(图4.4)。
- 为了准备测序,将SMRTbell模板与测序引物退火,随后一个DNA聚合酶分子结合到模板/引物结构上。
- 模板-引物-聚合酶复合物然后在测序前被固定到SMRT细胞的底部。
para
- 目前可用的PacBio SMRT测序仪(Sequel II/IIe)有两种测序模式,称为连续长读(CLR)和环形共识测序(CCS)。
- 在CLR模式下,DNA聚合酶沿着模板继续前进,直到停止,从而在一次过程中产生长读。
- 在CCS模式下,DNA聚合酶多次通过SMRTbell结构,并遍历模板的两条链,以生成共识读数(图4.4)。
4.2.2.3 Error Rate, Read Length, Data Output, and Cost
4.2.2.3 错误率、读取长度、数据输出和成本
para
- CCS模式显著提高了测序准确性。例如,在模板上进行十次通过后,准确性可以达到99.9%,即Q30。
- 在PacBio术语中,通过CCS模式生成的读段是高保真(或HiFi)读段。
- 相比之下,CLR读段的准确性较低,大约为90%。
- 一般来说,SMRT测序中最常见的错误类型是插入缺失(indels),其中大多数发生在同聚物区域。
- 尽管CCS读段更准确,但它们相对较短,目前范围在15-25 kb。
- 相比之下,CLR读段的长度通常为30-60 kb,据报道最大长度超过200 kb。
- 在数据输出方面,CLR在一次运行中生成150-250 Gb的数据,而CCS产生10-30 Gb。
- 根据PacBio测序试剂的当前列表价格,CLR读段的每Gb成本接近Illumina高端测序仪,而CCS读段由于需要多次通过以生成共识序列,成本仍然更高(见表4.1)。
4.2.2.4 Sequence Data Generation
4.2.2.4 序列数据生成
para
- 原始数据处理的目的是生成原始碱基调用,这一过程在测序仪上进行。
- 这包括处理电影以提取测序信号,从提取的轨迹和脉冲中进行碱基调用,以及对碱基调用的质量检查。
- 为了生成CLR读数,需要从原始聚合酶生成的读数中修剪适配器序列。
- 为了生成CCS读数,同样需要首先移除适配器序列以生成子读数。
- 每个子读数包含的序列对应于从一个方向对DNA模板的一次通过。
- 子读数的后续后处理将多个连续的子读数合并,以创建一个共识序列。
- 所有这些数据生成步骤都在测序仪上进行。
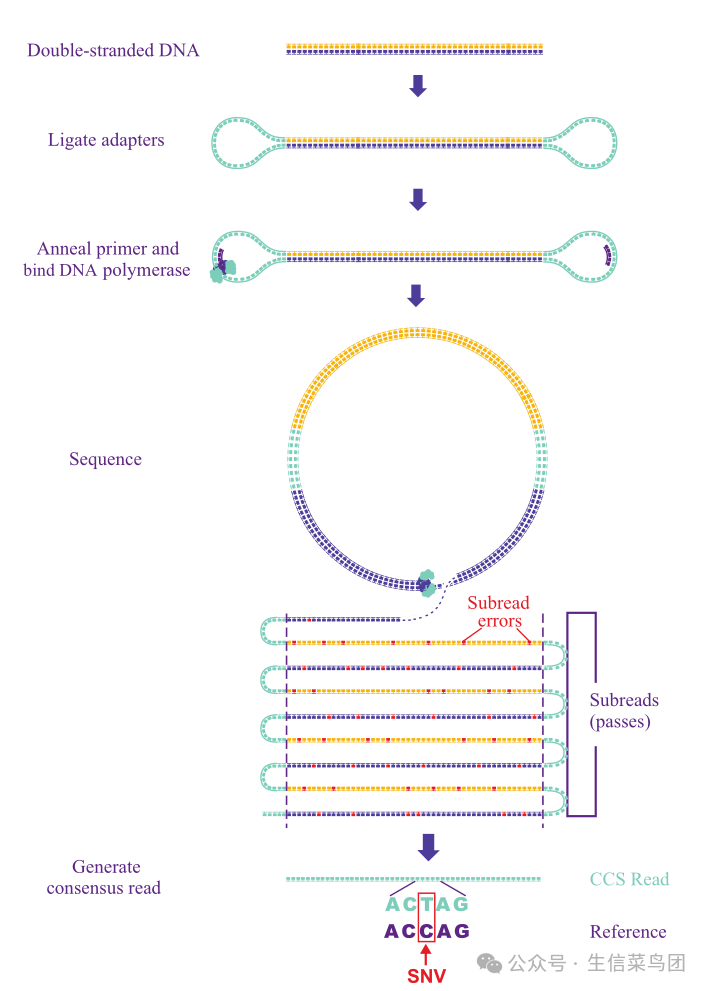
image-20241028011111687
- 图 4.4 PacBio 测序文库制备和测序。文库制备过程主要涉及将发夹环适配器连接以创建 SMRTbell 结构。SMRTbell 模板可以通过循环共识测序(CCS,如图所示)或连续长读段(CLR)模式进行测序。在 CCS 模式下,模板经历多次通过,每次通过产生易出错的子读段,随后生成准确的共识读段。(经许可改编自 Springer Nature 客户服务中心 GmbH: Springer Nature, Nature Biotechnology, 准确的循环共识长读段测序提高了变异检测和人类基因组的组装,Aaron M. Wenger 等人,版权 2019。)
4.2.3 Oxford Nanopore Technologies (ONT) Long-Read Sequencing
4.2.3 牛津纳米孔技术(ONT)长读长测序
4.2.3.1 Sequencing Principle
4.2.3.1 序列化原则
para
- 与 PacBio 平台类似,Oxford Nanopore 平台也是一个单分子测序平台,但它产生的读长可以更长。
- 它不是通过合成测序,而是通过物理测量,在分子通过生物纳米孔时检测 DNA(或 RNA)链上的核苷酸顺序(图 4.5)。
- 为了实现检测,首先在孔的两侧施加电压,该孔嵌入浸在电解质溶液中的电绝缘膜中。
- 由于 DNA/RNA 带负电荷,电场驱动它们通过孔。
- 在此过程中,纳米孔作为生物传感器,检测由核苷酸通过引起的离子电流的细微变化。
- 由于不同的核苷酸在此过程中导致不同的变化模式,收集到的电信号随后通过碱基识别算法解码,以揭示底层的核苷酸序列。
- 需要注意的是,检测到的离子电流变化来自 DNA/RNA 链上的 5-9 个核苷酸(实际检测的核苷酸数量随纳米孔 ONT 版本的不同而变化),而不是单个核苷酸。
- 为了实现可靠的检测,DNA/RNA 链通过孔的速度很重要,因为它需要减慢到允许可靠记录电流变化的速率。
- 这种速度控制是通过位于孔口的马达蛋白实现的,且在撰写本文时的速度通常在每秒 200-450 个碱基的范围内,具体取决于版本。
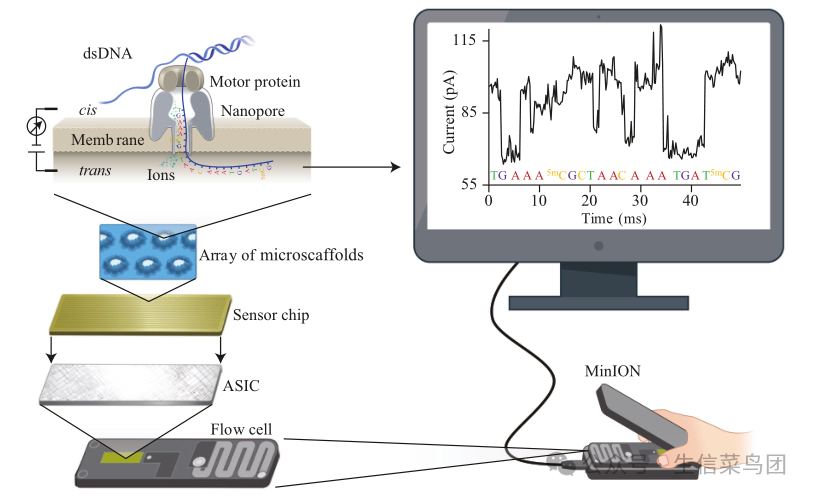
image-20241028011213420
- 图4.5 纳米孔测序。此处展示的是使用MinION流式细胞进行测序,该细胞包含512个通道,每个通道中有4个纳米孔。
- 携带纳米孔的绝缘膜由一个微支架阵列支撑,该阵列下方是一个传感器芯片。
- 传感器芯片上有与各个通道对应的电极,电极的电信号由专用集成电路(ASIC)记录。
- 记录的信号随后被分析以进行碱基识别。(经Springer Nature客户服务中心 GmbH:Springer Nature, Nature Biotechnology, 纳米孔测序技术、生物信息学和应用,Yunhao Wang等,版权2021年许可改编。)
4.2.3.2 Implementation
4.2.3.2 实施
para
- ONT目前提供三种主要设备,分别对应不同的数据吞吐量水平:MinION、GridION和PromethION。
- MinION位于低端,是一个USB驱动器大小的设备,容纳一个流动池。
- GridION作为升级版,可以容纳多达五个流动池。
- MinION和GridION使用的流动池是同一类型,包含2,048个纳米孔,分布在512个通道中。
- 高端的PromethION有能力同时运行多达48个流动池。
- PromethION使用的流动池容量更大,包含12,000个纳米孔,分布在3,000个通道中。
- 对于MinION/GridION和PromethION的流动池,每个通道中一次只能有一个孔进行测序。
- 除了这些流动池,对于常见的小规模测序,ONT还提供了一个名为Flongle的流动池适配器,它为MinION/GridION提供了一个适配器,允许使用更小、成本更低的流动池。
- Flongle适配器有126个通道,允许从126个纳米孔同时进行测序。
para
- ONT 提供两种测序模式,一种用于生成长读长(目前定义为低于100 kb),另一种用于超长读长(≥100 kb)。
- 输入 DNA 的长度决定了使用哪种模式。
- 长读长测序的样本文库制备过程包括片段化以及/或大小选择(可选)、末端修复、A-加尾和测序接头的连接。
- 超长测序文库制备需要提取超高分子量 DNA。
- 虽然超长测序文库制备的步骤可能会继续演变,但目前的程序包括一个转座步骤,该步骤同时切割模板并将标签连接到切割的末端,接下来的步骤是将测序接头添加到标记的末端,最后在将 DNA 文库加载到流动池之前进行过夜洗脱。
4.2.3.3 Error Rate, Read Length, Data Output, and Cost
4.2.3.3 错误率、读取长度、数据输出和成本
para
- ONT平台有能力对提交给它的DNA/RNA片段进行全长测序。
- 迄今为止,ONT实现的最长读取长度超过4 Mb。
- 通常情况下,长读测序模式的长度为10–100 kb,而超长读测序的长度为100–300 kb。
- 在撰写本文时,最新的纳米孔(R10.4)的原始读取错误率为1%,以达到99%(Q20)的准确性。
- 同聚物错误是ONT测序中最常见的错误类型。
- 在数据输出方面,MinION/GridION通常可以从每个流动池产生10–20 Gb的数据(在当前250个碱基/秒的移动速度下最多30 Gb)。
- PromethION的每个流动池的吞吐量为50–100 Gb(最多170 Gb)。
- 凭借最高装载容量为48个流动池,PromethION的数据输出可以超过PacBio Sequel II和Illumina NovaSeq 6000。
- 在MinION/GridION平台上测序的成本为每Gb US13–40。
- 同样,这一计算基于撰写本文时的每个流动池的标价除以每个平台的典型数据输出。
4.2.3.4 Sequence Data Generation
4.2.3.4 序列数据生成
para
- ONT 使用其 MinKNOW 软件进行设备控制、原始数据收集和数据处理以生成碱基调用。
- 为了进行板载实时碱基调用,使用其专有算法 Guppy。
- 与 Illumina 或 PacBio 平台相比,这些平台的碱基调用更为成熟,而来自纳米孔测序信号的碱基调用仍在由 ONT 和独立团队不断改进,并已取得显著进展。
- ONT 测序仪将收集的原始电信号存储在 FAST5 文件中,并可以在碱基调用后在 FASTQ 文件中提供序列读数。
- FAST5 文件中的原始数据可以用于独立碱基调用、DNA/RNA 碱基修饰的调用(例如,甲基化),以及使用其他软件工具进行下游分析。
- 由于序列读数的准确性在很大程度上依赖于所使用的碱基调用算法,随着时间的推移,对 FAST5 文件中原始数据的重新处理可能会导致新信息的发现。
- 然而,存储原始数据文件确实会显著增加对存储空间的需求。
4.2.4 Ion Torrent Semiconductor Sequencing
4.2.4 离子激流半导体测序
4.2.4.1 Sequencing Principle
4.2.4.1 序列化原则
para
- 在ONT平台之前开发的Ion Torrent半导体测序系统是第一个不依赖于化学修饰核苷酸、荧光标记和耗时图像扫描步骤的NGS平台,从而实现了更快的速度、更低的成本和更小的设备占地面积。
- Ion Torrent平台通过检测在测序合成过程中每个核苷酸掺入后释放的H⁺离子来测序DNA。
- 当一个核苷酸被掺入到新的DNA链中时,由DNA聚合酶催化的化学反应会释放一个焦磷酸盐基团和一个H⁺(质子)。
- H⁺的释放导致反应附近pH值的变化,这种变化可以被检测并用于确定上一个循环中掺入的核苷酸。
- 由于pH值的变化不是核苷酸特异性的,为了确定DNA序列,每种底物核苷酸(dATP、dGTP、dCTP和dTTP)按顺序在不同时间添加到反应中。
- 在引入核苷酸后检测到的pH变化表明模板链在最后一个位置包含其互补碱基。
4.2.4.2 Implementation
4.2.4.2 实施方案
para
- 该技术中的文库构建过程与其他NGS技术相似,涉及将平台特定的引物连接到DNA鸟枪法片段上。
- 然后,文库片段通过乳液PCR在3微米直径的珠子表面进行克隆扩增。
- 涂有扩增序列模板的微珠随后被沉积到Ion芯片中。
- 每个Ion芯片都有一个液体流动室,允许原生核苷酸(一次引入一个)的流入和流出,以及测序合成过程中所需的DNA聚合酶和缓冲液。
- 为了测量每次引入核苷酸可能引起的pH变化,芯片底部通过采用半导体行业标准工艺制造了数百万个pH微传感器。
4.2.4.3 Error Rate, Read Length, Data Output, and Cost
4.2.4.3 错误率、读长、数据输出和成本
para
- Ion Torrent平台的总体准确率超过99%。
- 主要的错误类型是由同聚物引起的插入或缺失。
- 当DNA模板包含同聚物区域时,即一段相同的核苷酸(例如TTTTT),pH变化的信号更强,并且与同聚物中包含的核苷酸数量成比例。
- 例如,如果模板包含两个T,dATPs的流入将产生大约是单个T产生的pH变化信号的两倍。
- 相应地,3个T的信号将是2个T的1.5倍,6个T的信号将减少到5个T的1.2倍。
- 因此,随着重复碱基总数的增加,信号强度比率逐渐降低,这降低了正确调用碱基总数的可靠性。
- 据估计,目前调用5个碱基同聚物的错误率为3.5%。
para
- 目前(截至2022年初)Ion Torrent家族中有三种测序系统:GeneStudio S5、Genexus和PGM Dx。
- 在这些系统中,GeneStudio S5可以产生长达600个碱基的读长,每片芯片的总输出量可达25 Gb。
- 另一方面,Ion PGM Dx系统生成200个碱基的读长,总输出量最多可达1 Gb。
- Genexus系统不仅仅是一个测序仪,它提供了一个从核酸提取、文库制备、模板准备到测序和报告的集成工作流程。
- Genexus使用其GX5芯片进行测序,表4.1中列出的成本仅适用于测序步骤。
- GeneStudio S5使用五种芯片类型:Ion 510、520、530、540和550,其中550芯片产生的数据最多(20-25 Gb),最具成本效益,而510芯片产生的数据最少(0.3-1 Gb),成本效益最低。
- PGM Dx系统使用的Ion 318 Dx芯片,吞吐量类似于510芯片,生成600 Mb到1 Gb的数据,适合运行不需要大量数据的分子诊断测试。
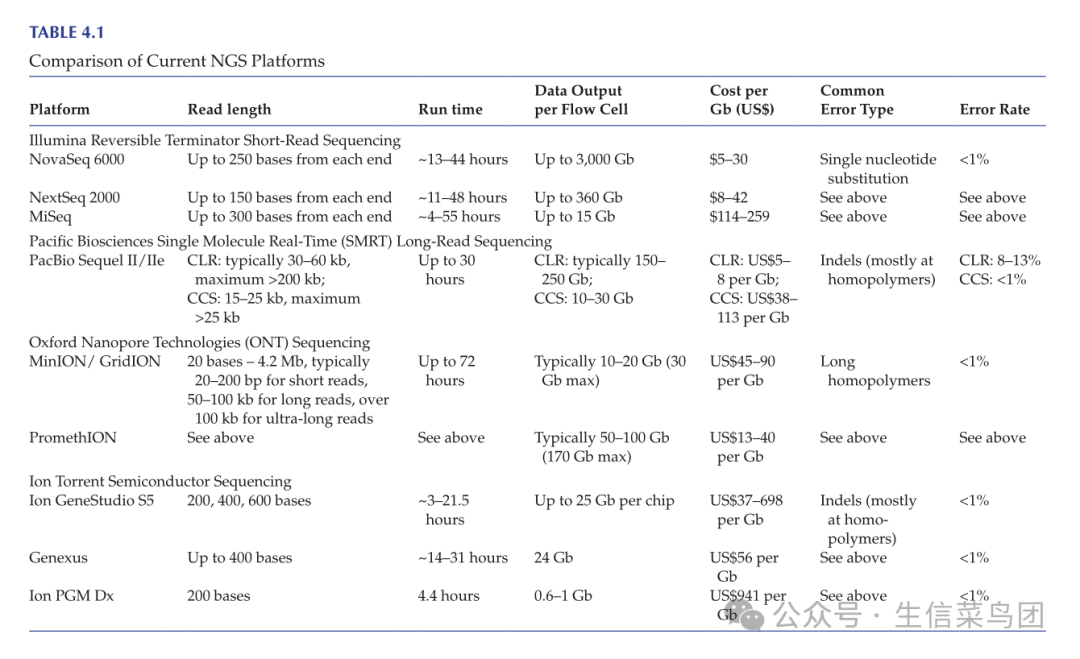
image-20241028011317056
4.2.4.4 Sequence Data Generation
4.2.4.4 序列数据生成
para
- GeneStudio S5 和 Ion PGM Dx 的测序器操作和碱基识别由 Torrent Suite Software 提供。
- Genexus 系统拥有自己的软件来管理工作流程并生成碱基识别。
- 软件使用的碱基识别过程相似。
- 来自 pH 变化的原始电压测量信号首先保存在每个循环的 DAT 文件中。
- 运行结束后,所有 DAT 文件被压缩成一个单一的 WELLS 文件,该文件随后用作生成碱基识别的输入。
- WELLS 文件可以保存并用于重新分析。
- 碱基识别结果报告在一个未映射的 BAM 文件中。
- 除了生成原始序列读数外,软件还提供额外的分析功能,包括读数修剪和过滤、读数映射到参考基因组,以及通过插件进行其他三级分析。
4.3 A Typical NGS Workflow
4.3 典型的下一代测序工作流程
para
- 尽管不同NGS技术在原理上存在差异,但整体的NGS工作流程是相似的。
- 使用这些NGS技术对基因组DNA或RNA转录物进行测序都涉及多个步骤(图4.6)。
- 这一过程的早期步骤是从感兴趣的生物样本中提取DNA或RNA分子,构建测序文库。
- 由于这些分子通常太大,无法直接被大多数NGS技术处理,尤其是那些产生短读长的技术,因此提取的DNA或RNA分子通常需要首先被分解成更小的片段。
- 这种片段化可以通过不同的技术实现,包括酶处理、声波剪切、超声处理或化学剪切(通常用于RNA)。
- 片段化步骤通常之后会进行一个大小选择步骤,以收集特定目标范围内的片段。
- 如果需要从长读长平台获得超长读长,则需要特殊的提取程序来保留高分子量DNA。
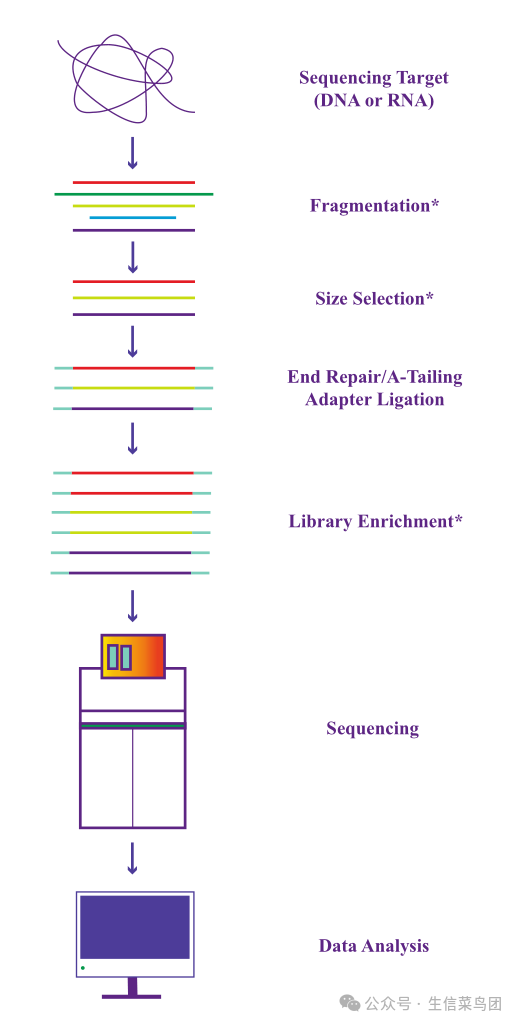
image-20241028011428568
- 图 4.6 NGS 实验的通用工作流程。在文库构建中,仅显示了不同测序平台共享的核心步骤。带星号的步骤在某些文库构建协议中不使用。连接到测序目标的适配器是特定于每个平台的。还有其他一些文库构建策略或程序,例如非连接或目标序列捕获,这里未显示。
para
- 测序文库构建过程中的一个关键步骤是将适配器连接到DNA片段的两端。
- 对于RNA片段,通常先将它们转化为互补DNA(cDNA),然后再添加适配器。
- 适配器是人工序列,包含多个组件,包括在各个片段上启动测序反应的通用测序引物序列,以及用于在多个样本一起测序时区分它们的索引(或"条形码")序列。
- 虽然它们在不同NGS平台中通常具有相似的功能,但实际的标准化适配器序列是特定于每个平台的。
- 只要平台的关键适配器序列元素到位,也可以设计定制适配器以满足特殊需求。
- 在适配器连接之前,需要通过末端修复步骤准备DNA(或cDNA)片段的两端。
- 适配器连接后,结果文库中的测序DNA模板可能需要通过PCR扩增步骤使用适配器中的常见序列进行富集。
- 或者,构建的文库可以在大多数平台上无需富集进行无PCR测序。
4.4 Biases and Other Adverse Factors That May Affect NGS Data Accuracy
4.4 影响NGS数据准确性的偏见和其他不利因素
para
- 正如一定水平的错误碱基调用是NGS平台固有的,导致序列调用生成的多个步骤也并非没有偏差。
- 与错误不同,偏差会影响原始DNA或RNA群体的准确表示,导致某些序列的代表性高于(或低于)预期。
- NGS中偏差的主要来源是文库构建和测序过程本身所涉及的分子步骤。
- 除了偏差,还有其他潜在因素可能导致生成不准确的测序信号。
- 以下是测序文库构建和测序过程中可能影响NGS数据准确性的各种潜在偏差和其他不利因素的详细说明。
- 需要注意的是,虽然无法完全避免这些因素,但意识到它们的存在是通过仔细的实验设计和数据分析以及开发更稳健的分析算法来最小化其影响的第一步。
4.4.1 Biases in Library Construction
4.4.1 文库构建中的偏差
para
- DNA断裂和片段大小选择的偏差。文库构建的初始步骤,即DNA断裂,通常被认为是随机过程,不依赖于序列背景。
- 然而,这已经被证明并非如此。
- 例如,声波处理和雾化处理导致在C残基后发生DNA链断裂的频率高于预期。
- 在DNA断裂后,大小选择过程也可能引入偏差。
- 例如,如果使用凝胶提取进行这一过程,高凝胶熔点温度的使用有利于回收高GC含量的片段。
para
- 连接偏倚。在片段化和大小选择之后,双链DNA片段通常会在末端修复后,在两个3’-端进行腺苷酸化,生成3’-dA尾,以促进后续携带5’-dT突出端的适配器连接,从而避免DNA片段或适配器的自连接。
- 然而,这种基于AT突出端的适配器连接过程,往往对以T开头的DNA片段存在偏倚。
- 大型RNA物种的测序,如mRNA或长链非编码RNA,也受到这种偏倚的影响,因为从这些物种反转录而来的cDNA分子也要经历相同的适配器连接过程。
- 小RNA测序不受这种偏倚的影响,因为在小RNA测序文库制备中,适配器的连接是在反转录步骤之前进行的。
- 然而,小RNA适配器连接步骤引入了另一种类型的偏倚,这种偏倚以序列特异性的方式影响一些小RNA。
- 序列特异性是小RNA二级和三级结构的基础,这也受到连接反应混合物中温度、阳离子浓度和去稳定有机试剂(如DMSO)的影响。
- 小RNA适配器连接的效率受其二级和三级结构的影响。
para
- PCR偏差。在适配器连接后,DNA文库通常通过PCR在大多数当前NGS平台上进行富集测序。
- 基于DNA聚合酶的使用,PCR已知对极度富含GC或AT的DNA片段存在偏差。
- 这可能导致不同基因组区域的覆盖度变化,以及富含GC或AT区域的代表性不足。
- 虽然优化PCR条件可以在一定程度上减轻这种偏差,尤其是对于高GC区域,但这种偏差只能通过采用无PCR的工作流程来消除。
- 为此,Illumina提供了无PCR的选项。
- 对于在PacBio SMRT和ONT平台上进行的单分子测序,通常不需要PCR扩增,除非起始DNA/RNA的输入量较低。
4.4.2 Biases and Other Factors in Sequencing
4.4.2 序列中的偏差和其他因素
para
- 与PCR类似,大多数当前NGS系统所进行的合成测序过程也基于DNA聚合酶的使用,这引入了相似的覆盖偏差,对极端GC或AT含量的基因组区域不利。
- 由于DNA聚合酶的使用是这些技术的核心,因此很难完全消除这种偏差。
- 然而,在测序极端高GC或AT含量(>90%)的基因组或基因组区域时,应牢记这种偏差。
- 除了这种酶促过程外,测序过程中的其他方面,包括设备操作和调整、图像分析以及碱基识别,也可能引入偏差和伪迹。
- 例如,缓冲液中的气泡、晶体、灰尘和绒毛可能会遮蔽现有的簇(或珠子),导致产生人工信号。
- 扫描阶段的错位,甚至无意中的光线反射,都可能导致显著的成像不准确。
- 与上述一些固有的偏差不同,这些伪迹可以通过经验丰富的人员来最小化或避免。
para
- 测序信号处理和碱基识别步骤也可能引入偏差。
- 例如,在Illumina平台上,碱基识别结果可能受到颜色串扰、空间串扰以及相位和预相位的影响。
- 在MiSeq上,例如,每个循环后从四个检测通道生成四幅图像,这些图像需要叠加以提取用于碱基识别的信号强度。
- 这一过程因三个因素而复杂化:1)来自四个通道的信号并不完全独立,因为A和C通道之间以及G和T通道之间存在串扰,这是由于它们荧光标签的发射光谱重叠所致。
- 2)相邻的簇可能部分重叠,导致相邻簇之间的空间串扰。
- 3)来自特定循环的信号也依赖于之前和之后循环的信号,由于相位和预相位的影响。
- 虽然Illumina的专有软件在处理这些因素进行碱基识别方面效率很高,但还有其他商业和开源工具采用不同的算法来完成这些任务,并生成不同的结果。
- 这些方法使用的算法(包括Illumina方法)对信号分布做出了不同的假设,这些假设可能并不严格代表收集到的数据,因此会引入方法特定的偏差到碱基识别中。
4.5 Major Applications of NGS
4.5 NGS的主要应用
4.5.1 Transcriptomic Profiling (Bulk and Single-Cell RNA-Seq)
4.5.1 转录组分析(批量及单细胞RNA测序)
para
- NGS 已取代微阵列成为检测转录组谱和变化的主要手段。
- 生物样本(如细胞、组织或器官)的转录组谱由其发育阶段、内部和外部功能状态决定并反映其状态。
- 通过测序转录组中现有的 RNA 种类,NGS 提供了对关键问题的答案,例如哪些基因是活跃的以及其活跃程度。
- 单细胞水平的 RNA-seq 探究细胞异质性,揭示混合细胞群体或组织中的不同细胞类型和状态。
- 转录组研究几乎总是比较研究,对比一种组织/阶段/条件与另一种。
- 除了基因水平分析外,RNA-seq 还可以用于研究通过可变剪接来自同一基因的不同转录本。
- 作为转录组的一部分,小 RNA 也可以通过 NGS 进行类似研究。
- 来自大 RNA 和小 RNA 种类的批量 RNA-seq 数据分析分别在第 7 章和第 9 章中介绍。
- 由于其独特性,单细胞 RNA-seq 数据分析在专门的章节(第 8 章)中介绍。
4.5.2 Genetic Mutation and Variation Identification
4.5.2 遗传突变与变异识别
para
- 在人群中检测和编目个体间的基因突变或变异是下一代测序(NGS)的主要应用。
- 现有的NGS研究已经表明,像癌症和自闭症这样的严重疾病与新的体细胞突变有关。
- 诸如千人基因组计划之类的项目已经揭示了人群中大量的基因变异,这些变异解释了个体在物理特征、疾病易感性和药物反应上的差异。
- 第10章重点介绍了在研究环境中用于识别突变和各种类型变异的数据分析技术,并测试它们与特征或疾病的关联。
- 第11章重点介绍了NGS在临床应用中识别可操作变异以指导床边决策的方法。
4.5.3 De Novo Genome Assembly
4.5.3 新生基因组组装
para
- 桑格测序曾被认为是从头基因组组装的金标准,但越来越多的基因组,包括大型复杂基因组,已经仅通过NGS读段组装完成。
- NGS领域的技术进步,包括短读技术读长度的逐渐增加和长读技术的成熟,都促成了这一趋势。
- 基于NGS的基因组组装新算法的发展是这一进步背后的另一股力量。
- 第12章重点介绍了如何使用这些算法从NGS读段组装新基因组。
4.5.4 Protein-DNA Interaction Analysis (ChIP-Seq)
4.5.4 蛋白质-DNA相互作用分析(ChIP-Seq)
para
- 基因组的正常功能依赖于其与多种蛋白质的相互作用。
- 例如,转录因子是已知与DNA相互作用的蛋白质之一。
- 许多这类蛋白质以序列或区域特异性的方式与DNA相互作用。
- 为了确定这些蛋白质结合到基因组的哪些区域,可以通过一种称为染色质免疫沉淀(ChIP)的过程首先捕获结合区域,然后通过NGS进行测序。
- ChIP-seq可以应用于研究某些条件,如发育阶段或疾病,如何影响蛋白质因子对其亲和区域的结合。
- ChIP-seq数据分析在第13章中有所涵盖。
4.5.5 Epigenomics and DNA Methylation Study (Methyl-Seq)
4.5.5 表观基因组学与DNA甲基化研究(甲基化测序)
para
- 某些核苷酸和组蛋白的化学修饰为基因组调控提供了除嵌入在基因组初级核苷酸序列中的调控机制之外的额外层次。
- 这些修饰及其提供的调控信息构成了表观基因组。
- 基于NGS的表观基因组学研究揭示了同卵双胞胎在某些表型上的差异,以及表观基因组图谱的变化可能导致癌症等疾病。
- 胞嘧啶甲基化是表观基因组变化的主要形式。
- 第14章涵盖了DNA甲基化测序数据的分析。
4.5.6 Metagenomics
4.5.6 元基因组学
para
- 要研究像肠道微生物组或一桶海水中存在的微生物群落,其中存在极其庞大但未知的物种数量,一种涉及研究该群落中所有基因组的暴力方法就是宏基因组学。
- 最近,宏基因组学领域因下一代测序(NGS)技术的发展而得到了极大的推动。
- 通过快速测序宏基因组中的所有内容,研究人员可以获得微生物群落的组成和功能状态的全面概况。
- 与从单个基因组生成的NGS数据相比,宏基因组学数据要复杂得多。
- 第15章重点介绍了宏基因组学NGS数据分析。
5 Early-Stage Next-Generation Sequencing (NGS) Data Analysis: Common Steps
para
- 总的来说,NGS数据分析分为三个阶段。
- 在初级分析阶段,基于测序过程中产生的光学或理化信号的解卷积来调用碱基。
- 无论测序平台或应用如何,碱基调用结果通常存储在标准的FASTQ格式中。
- 每个FASTQ文件包含大量读数,即从测序文库中采样得到的DNA片段的序列读数。
- 在次级分析阶段,FASTQ文件中的读数进行质量检查、预处理,然后映射到参考基因组。
- 数据质量检查或控制(QC)步骤涉及检查一系列序列读数的质量指标。
- 基于数据QC结果,NGS测序文件进行预处理,以过滤掉低质量读数,修剪掉读数中低质量碱基调用的部分,并移除适配器序列或其他人工序列(如PCR引物,如果存在的话)。
- 随后将预处理后的读数映射(或对齐)到参考基因组,旨在确定读数来自基因组的哪个位置,这是大多数三级分析(除从头基因组组装外)所需的关键信息。
- 三级分析阶段高度依赖于具体应用,并在第三部分各章节中详细阐述。
- 本章重点介绍初级和次级阶段的步骤,特别是读数QC、预处理和映射,这些步骤在大多数应用中是常见且共享的(图5.1)。
5.1 Basecalling, FASTQ File Format, and Base Quality Score
5.1 基因序列读取、FASTQ文件格式及碱基质量分数
para
- 初级阶段从荧光图像、电影或物理化学测量中进行碱基识别的过程是通过平台特定的专有算法进行的。
- 例如,Illumina 使用基于统计模型的碱基识别器 Bustard。
- ONT 目前使用的是基于深度学习方法的 Guppy,该方法称为 RNN(或循环神经网络)。
- 除了这些随测序仪附带的内置碱基识别器外,还开发了其他独立的碱基识别算法,以进一步提高准确性。
- 提高碱基识别的准确性对于长读长测序平台尤为重要,ONT 平台就是一个很好的例子,其碱基识别算法正在积极开发中。
- ONT 开发了多种基于机器学习的碱基识别器,包括 Albacore、Scappie、Flappie 和 Bonito,除了 Guppy。
- 在撰写本文时,Guppy 的速度比其他碱基识别器更快,同时保持了相对较高的准确性。
- 而 Bonito 作为 ONT 最新一代的碱基识别器,使用了另一种深度学习方法 CNN(卷积神经网络),以实现比 Guppy 更高的准确性,但速度较慢。
- 社区开发的其他开源碱基识别算法包括 DeepNano、Nanocall、Chiron 和 causalcall。
- 由于这些算法开发工作的结果,碱基识别的准确性已取得了显著进步。
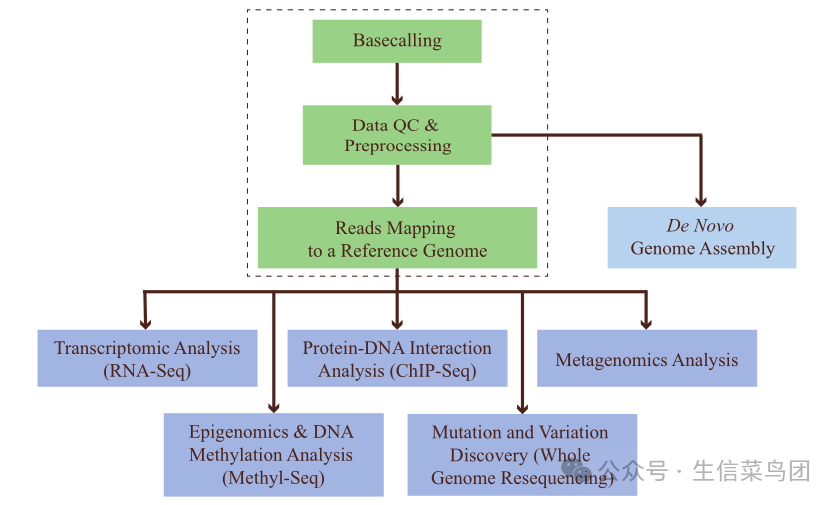
image-20241028013519497
- 图5.1 NGS数据分析概述。虚线框中的步骤是初级和次级分析中常见的步骤。
para
- 大多数终端用户通常不会干预碱基识别过程,而是专注于分析碱基识别结果。
- 无论测序平台如何,碱基识别结果通常以普遍接受的FASTQ格式报告。
- 在文件大小方面,一个典型的压缩FASTQ文件通常在多个GB范围内,可能包含数百万到数十亿个读取数据。
- 简而言之,FASTQ格式是一种基于文本的格式,包含每个读取的序列以及每个碱基的置信度评分。
- 图5.2展示了以FASTQ格式报告的这样一个读取序列的示例。

image-20241028013424951
- 图5.2 FASTQ序列读取报告格式。此处展示的是从NGS实验生成的一个读取。FASTQ文件通常包含数百万到数十亿个这样的读取,每个读取包含几行,如下所示。
- 第1行,以符号‘@’开头,包含序列ID和描述符。
- 第2行是读取序列。
- 第3行(可选)以‘+’符号开头,其后可能跟有序列ID和描述。
- 第4行列出了读取序列(第2行)中每个对应碱基的置信度(或质量)分数。
- 对于Illumina生成的FASTQ文件,此例中第1行的序列ID基本上标识了序列生成的位置。
- 这些信息包括设备(上述示例中的"HISEQ")、序列运行ID("131")、流动池ID("C5NWFACXX")、流动池泳道("1")、泳道内的瓦片编号("1101")、瓦片内序列簇的x/y坐标(分别为"3848"和"2848")。
- 随后的描述符包含有关读取编号的信息(此处"1"表示单次读取;对于配对末端读取,它可以是1或2)、读取是否被过滤(此处"N"表示未过滤)、控制编号("0")和索引(或样本条形码)序列("CGAGGCTGCTCTCTAT")。
para
- 置信度(或质量)评分,作为衡量错误碱基调用概率的指标,是FASTQ格式的一个基本组成部分。
- NGS碱基调用质量评分(Q评分)类似于Sanger测序中使用的Phred评分,其计算方式为:
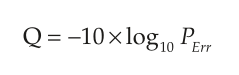
image-20241028013353684
para
- PErr 是发生碱基识别错误的概率。
- 基于这个公式,1%的错误识别碱基的几率相当于Q得分为20,而Q30意味着错误识别的几率为1/1000。
- 通常,一个碱基识别要可靠,其Q得分至少要达到20。
- 高质量的识别通常Q得分在30以上,通常达到40。
- 为了更好地可视化与相应碱基识别相关的Q得分,它们通常使用ASCII字符进行编码。
- 虽然存在不同的编码方案版本(例如,Illumina 1.0、1.3和1.5),但目前NGS领域大多已统一使用与Sanger测序相同的编码方案(图5.3)。
- 在图5.2所示的FASTQ示例中,第一个碱基C的编码Q得分为B,即33。
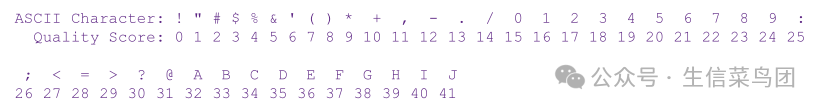
image-20241028013322055
- 图5.3 使用ASCII字符编码碱基质量分数。ASCII代表美国信息交换标准代码,ASCII码是计算机中字符的数值表示(例如,字母‘B’的ASCII码是66)。在这种编码方案中,ASCII字符码等于Q分数加33。当前主要的下一代测序(NGS)平台,包括Illumina(1.8版本之后),都使用这种编码方案来表示Q分数。
para
- 为了计算PErr,通常在Illumina测序中使用控制泳道或spike控制来生成一个基础调用分数校准表以供查找。
- 在没有控制泳道和spike控制的情况下,也可以使用预先计算的校准表。
- 由于每个平台对Q分数的校准方式不同,如果需要相互比较或以集成方式分析,它们的Q分数需要进行重新校准。
- 为了进行重新校准,使用一组映射到参考基因组中没有SNP区域的读段,读段与参考序列之间的任何不匹配都被视为测序错误。
- 基于读段中每个碱基位置的错配率,构建一个新的校准表,然后用于重新校准。
- 即使不进行跨平台NGS数据比较和整合,同一平台上生成的NGS数据在映射后仍可以使用相同的方法进行重新校准(见下文),这通常会导致基础调用质量分数的提高。
5.2 NGS Data Quality Control and Preprocessing
5.2 NGS 数据质量控制和预处理
para
- 在NGS数据生成后,第一步应该是数据质量检查。
- 虽然这一步骤并不直接生成生物学见解,但它仍然是必不可少的,并且应该仔细进行。
- 这样做可以避免在后续步骤中产生无意义甚至错误的结果,以及不必要的计算资源和时间的消耗。
- 在此过程中,需要检查以下数据质量指标:
-
- Q分数:这些可以通过不同方式进行检验。
- 在单碱基基础上,可以通过检查所有读数的所有碱基位置的质量分数来进行,从第一个测序碱基到最后一个。
- 作为一个普遍趋势,对于基于合成测序的平台,在测序过程的早期阶段覆盖的碱基位置往往比后期测序的碱基位置具有更高的Q值。
- 然而,即使是后期碱基位置的Q分数,其平均值也应至少为20。
- 如果在后期出现显著的Q分数下降,受影响的碱基位置需要仔细检查,并且应从受影响的读数中剪掉低质量的碱基。
- 此外,N调用的增加百分比也有助于确定碱基调用质量的损失(当碱基调用算法无法自信地调用任何四种碱基时,称为N)。
- 另一种检查Q分数的方法是绘制每个读数的平均Q分数,并检查其分布模式。
- 对于一次成功的运行,大多数读数的平均Q分数应超过30,只有极少数读数的平均Q分数低于20。
-
- 每个位置每种碱基的百分比:如果读数来自由随机生成的DNA片段构建的测序文库,那么在每个碱基位置观察到四种碱基中每一种的几率应该是恒定的。
- 因此,当绘制所有碱基位置每种碱基的百分比时,A、C、G和T的图应该大致平行,并且每个图中显示的总百分比应反映目标文库中每种碱基的总体频率。
- 如果图形显著偏离平行,这表明文库构建过程中存在问题,例如文库中存在过度代表性的序列(如RNA-seq文库中的rRNA),或非随机片段化。
-
- 读数长度分布:对于产生不同长度读数的平台(如PacBio和ONT平台),也应检查读数长度的分布。
- 结合Q分数的分布,这决定了运行生成的有用数据的总量。
- 此外,在数据质量和总体量相等的情况下,产生更长读数的运行在序列比对或组装方面比产生更多相对短读数的运行更有优势。
para
- 除了检查读段的质量和长度分布外,还应检查其他质量控制指标,例如是否存在人工序列,包括适配器和PCR引物,或基于序列同一性的重复序列(序列重复也可以基于参考基因组映射结果进行检查)。
- 在检查序列数据质量后,应进行过滤以去除低质量读段。
- 此外,如果存在低质量的碱基调用,例如3'端Q分数低于20的碱基,以及人工序列污染物,也应进行修剪。
- 虽然一些平台(例如Illumina)在生成FASTQ文件之前默认进行序列过滤,但如果检查后发现序列Q分数分布不令人满意,可能需要执行额外的过滤/修剪。
- 执行这些预处理任务是高质量下游分析的要求。
para
- 最常用的下一代测序(NGS)数据质量控制软件包括FastQC、NGS QC Toolkit和fastp。
- 这些工具包具有功能模块,用于检查每个读取和每个碱基的Q分数、碱基频率分布、读取长度分布,以及重复序列和人工序列的存在。
- FastQC是用Java编写的,在大多数操作系统(包括Windows)上具有用户友好的界面。
- fastp用C/C++开发,采用多线程进行并行处理,旨在实现快速的质量控制速度,以及其他预处理步骤,包括适配器修剪、质量过滤、每个读取的质量修剪等。
- 较新的NGS质量控制工具,如seqQscorer,应用机器学习方法,试图更好地理解质量问题并实现自动质量控制。
- 诸如FQC Dashboard和MultiQC之类的工具充当其他工具(例如FastQC)的QC结果聚合器,并将它们呈现在单个报告中。
- 数据质量控制后,为了执行独立的预处理任务,如适配器修剪和读取过滤,通常使用cutadapt和Trimmomatic等工具。
para
- 上述提到的一些质量控制工具,包括FastQC和fastp,可以用于短读和长读。
- 还有一些工具,如NanoQC(NanoPack的一部分),专门用于长读质量控制。
- 除了NanoQC,NanoPack还包含一系列用于修剪、过滤、总结、可视化等功能的实用工具。
- PycoQC是另一个为ONT数据提供交互式质量控制指标的工具。
- 对于PacBio长读,SequelTools提供质量控制数据,以及其他任务,如读段过滤、总结和可视化。
5.3 Read Mapping
5.3 读映射
para
- 数据清理后,下一步是将读段映射或对齐到参考基因组(如果可用),或者进行从头组装。
- 如图5.1所示,大多数NGS应用在进行进一步分析之前,需要将读段映射到参考基因组。
- 这个映射过程的目的是确定读段在基因组中的起源位置。
- 与使用BLAST等工具在基因组中搜索单个或少量序列的位置相比,将数百万个NGS读段(有时非常短)同时映射到基因组并非易事。
- 进一步的挑战来自于这样一个事实:由于多态性和突变,任何特定基因组(NGS读段所来源的)在许多位点上都偏离参考基因组。
- 因此,为这项任务构建的任何算法都需要能够容纳这种序列偏差。
- 更复杂的是,测序错误通常与真实的序列偏差难以区分。
5.3.1 Mapping Approaches and Algorithms
5.3.1 映射方法与算法
para
- 将NGS读段映射到参考基因组本身并不是一项新任务。
- 如上所述,在NGS出现之前,已经存在许多序列比对算法,其中最著名的是BLAST。
- 这些比对工具使用哈希表和种子扩展方法来执行将单个查询序列与序列数据库(如GenBank)比对的计算密集型过程。
- 然而,使用这些方法将数百万个NGS读段映射到参考基因组会引发可扩展性问题,因为它们无法扩展到数据量,也无法缩小到许多NGS读段的短长度(因为短读段携带的信息较少)。
- 因此,为了映射NGS读段,已经设计出了新一代的算法,要么通过优化先前的方法,要么引入新的方法。
para
- NGS读段的映射可以分为三个步骤(图5.4)。
- 第一步是在计算机内存中对参考基因组序列进行索引,以提高后续搜索速度。
- 使用哈希表是实现这一目标的一种方法,而另一种方法是使用基于后缀树的技术(接下来将详细介绍)。
- 第二步是所谓的"全局定位",其目标是确定从读段中提取的种子序列在已索引的参考基因组中的可能匹配位置。
- 第三步是读段整体与第二步中匹配的位置进行广泛对齐,以验证对齐,同时生成对齐信息,如序列变异及其类型。
para
- 基于哈希表的参考基因组索引通常用于快速查找读段中精确子序列(或k-mer)的匹配位置。
- 哈希表是一种存储关联数据的数据结构,在这种情况下,存储的是k-mer及其相关的基因组位置。
- 在基因组序列通过哈希进行索引后,从每个读段中提取k-mer,并将其用作种子在哈希表中搜索,以识别它们在基因组中的可能位置(图5.4和5.5)。
- 基于此方法的比对工具包括minimap2、SOAP(短寡核苷酸比对程序)、MAQ(带质量值的映射和组装)、Illumina的Isaac基因组比对软件以及Novoalign(商业软件)。
- 在这些比对工具中,minimap2使用最小化器来减少存储哈希表所需的计算机内存量,以进一步提高速度。
- 需要注意的是,虽然大多数比对工具使用哈希表来索引参考基因组,但有些比对工具(如MAQ)是从NGS读段中创建哈希表,在这种情况下,从参考基因组中提取的k-mer用于在读段中查找匹配项。
para
- 在BLAST使用的种子扩展方法中,所使用的种子是连续序列,旨在定位近似匹配,这对于包含变异特别是插入缺失的序列比对来说并不理想。
- 为了提高比对的鲁棒性,NGS读取比对器已经从使用连续精确匹配种子迁移到使用非连续(或间隔)种子。
- 通过允许种子之间有间隔,找到匹配的机会增加了。
- 例如,在SOAP和Novoalign中,为了使用间隔种子进行比对,首先将参考基因组序列切成等大小的小片段,并存储在内存中的一个大哈希表中。
- 然后,NGS读取以类似方式切成子序列,这些子序列与参考基因组进行比对(图5.5a)。
- 从计算角度来看,这些比对器对内存和处理器要求较高,因此速度并不快。
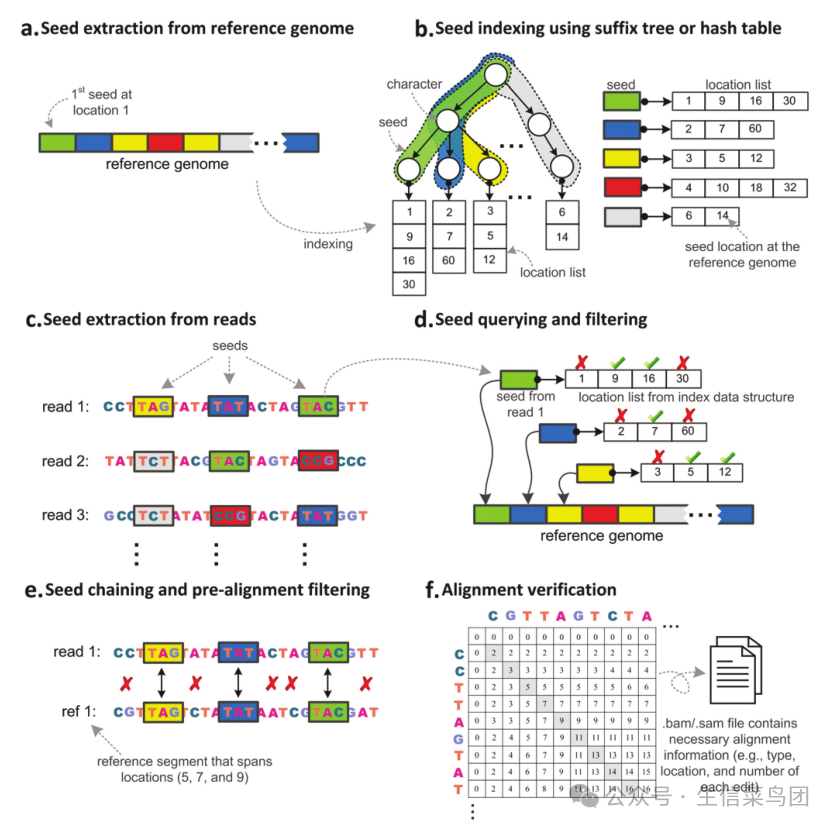
image-20241028014819479
- 图5.4 映射NGS读段的 主要步骤。在第一阶段,参考基因组被索引。这是通过从参考基因组中提取种子序列(a)并随后使用后缀树或哈希表对种子序列进行索引(b)来实现的。
- 在第二阶段,从读段中提取种子序列(c),然后用于搜索索引参考基因组中可能的匹配位置(d)。
- 在所示示例中,从读段1中提取的每个种子都被搜索以定位它们在索引基因组中的潜在位置。
- 根据它们的邻近性,一些位置被排除(红色X),因为这些位置不太可能跨越读段。
- 在最后阶段,相邻的种子被链式连接,种子之间的间隙序列被检查是否存在不匹配(红色X),基于此,预对齐过滤器确定是否接受读段与基因组区域之间的对齐(e)。
- 在最后一步,对齐结果需经过验证,以生成包括序列差异及其位置的 alignment results including sequence differences and their locations.
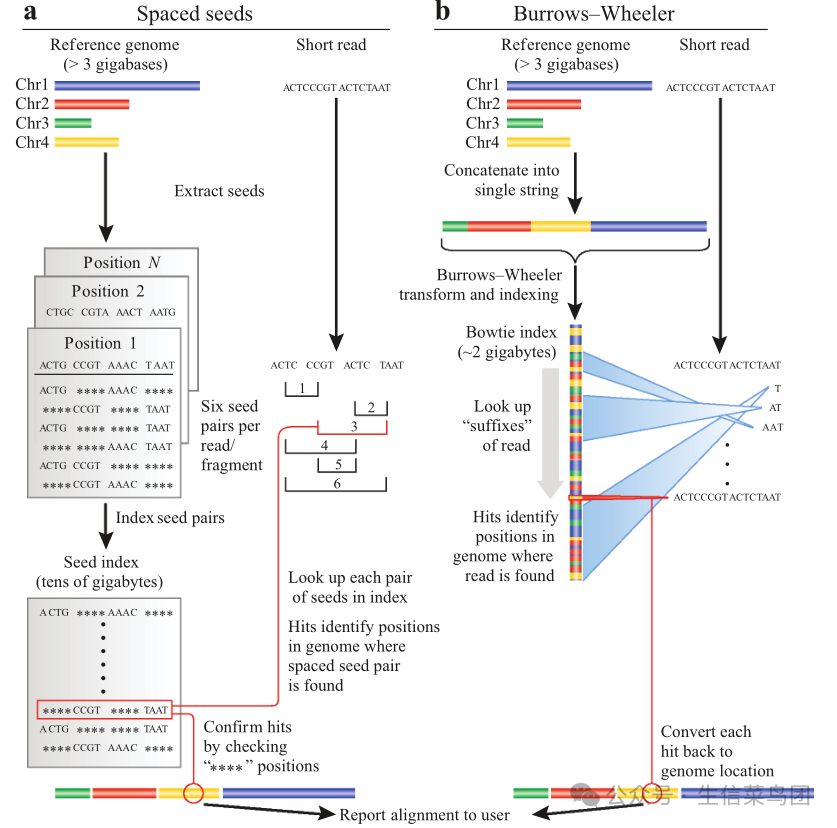
image-20241028014858904
- 图5.5 NGS读段映射方法。面板(a)展示了基于间隔种子索引的方法。在此图中,从参考基因组序列中提取的间隔种子通过哈希表进行索引。
- 面板(b)展示了基于Burrows-Wheeler变换(BWT)的方法。在此示例中,算法Bowtie通过逐个碱基查找读段,从右到左,与经过变换和索引的基因组进行映射。
para
- 为了减少对计算资源的需求,另一种方法是使用Burrows-Wheeler变换(或BWT)。
- BWT是一种算法,它对一块文本(在NGS数据的情况下是基因组核苷酸序列)进行可逆变换,以实现无损数据压缩。
- 这种变换通过文本重排过程实现,可以高效地利用后缀数组数据结构来实现(见图5.6a)。
- 变换后,可以使用Ferragina和Manzini提出的算法高效地索引参考基因组序列。
- 图5.6(b)提供了一个示例,展示了如何使用这种后缀数组在索引的基因组中查找k-mer。
- 结合使用BWT、后缀数组和FM索引,有效减少了存储索引参考基因组所需的计算机内存量,以实现快速映射。
- 例如,使用这种方法,索引的人类基因组仅占用2-3GB的计算机内存,而不是空间种子索引方法所用的超过50GB,并且运行时间从数小时缩短到数分钟。
- BWT被诸如BWA(Burrows-Wheeler对齐)[25]、Bowtie/Bowtie 2[26, 27]和SOAP2[28]等算法所采用。
- 图5.5b展示了这种方法在Bowtie案例中的工作示例。
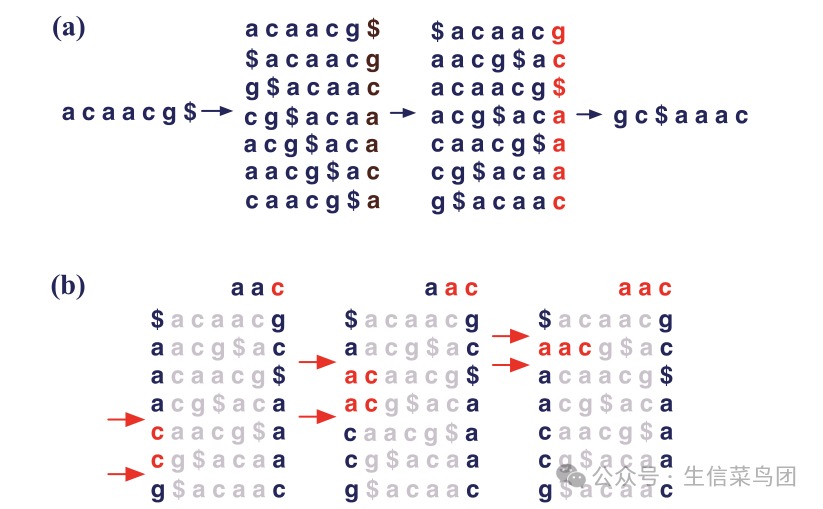
image-20241028015042038
- 图5.6展示了Burrows-Wheeler变换(BWT)如何用于对NGS读段进行比对。面板(a)显示了BWT过程的一个简短示例序列‘acaacg’。面板(b)说明了如何使用BWT来识别以‘aac’为前缀的读段序列的位置。(经Springer Nature客户服务中心GmbH:Springer Nature,基因组生物学,超快速且内存高效的短DNA序列与人类基因组比对,B Langmead, C Trapnell, M Pop, 和 SL Salzberg,版权2009年许可改编。)
para
- 在基于后缀树或哈希表的使用确定了NGS读数中种子序列的全局位置后,相邻的种子被链接起来,并评估它们之间的间隙以检查错配。
- 如果通过了这一过滤过程,并且读数和基因组区域匹配,那么接下来使用成对比对来验证对齐并生成SAM或BAM格式的对齐结果(见下文)(图5.4)。
- 这种成对比对过程可以通过不同的技术来执行,其中Smith–Waterman、Hamming Distance和Needleman–Wunsch算法经常被使用。
- Smith–Waterman算法在常用的工具如STAR中得到了应用。
- Smith–Waterman是一种基于动态规划(DP)的局部对齐程序。
- 动态规划最早由Needleman和Wunsch在1970年引入,用于DNA和蛋白质序列的对齐,目标是生成两个序列的最大对齐得分。
- 然而,Needleman–Wunsch算法本身用于全局对齐。
- 如Novoalign这样的对齐方法使用Needleman–Wunsch算法。
- Hamming Distance是一种非基于动态规划的不相似性度量,即两个序列中核苷酸不同的位置数。
- 基于HD的方法被RMAP、Bowtie和mrsFAST所使用。
5.3.2 Selection of Mapping Algorithms and Reference Genome Sequences
5.3.2 映射算法和参考基因组序列的选择
para
- 表5.1列出了一些常用的映射算法。
- 在选择比对工具时,需要考虑准确性、速度和计算机内存需求等因素。
- 由于这些因素通常是相互冲突的,一些比对工具更注重准确性,而另一些则强调速度和内存效率。
- 如果速度和内存效率更重要,推荐使用Bowtie2。
- 如果更偏好高准确性,通常使用基于哈希表的工具,如Novoalign、Stampy和SHRiMP2。
- BWA在速度和敏感性之间取得了平衡。
- 这些比对工具大多数最初是为了映射非常短的读取而开发的,例如早期Illumina测序仪的35个核苷酸读取。
- 随着读取长度的逐渐增加,这些比对工具也相应地进行了适应。
- 例如,BWA-MEM是对原始BWA算法的改进,用于比对更长的Illumina短读取。
image-20241028015139715
para
- 对于对齐更长的读段,例如来自PacBio和ONT平台的读段,应使用设计用于处理长序列的对齐工具,如minimap2、GraphMap、BLASR、LAST、NGMLR或Winnowmap/Winnowmap2。
- 通常,长读段的映射遵循与短读段对齐器类似的种子-链-扩展方法。
- 为了有效映射,长读段通常被分解成较短的子序列,这些子序列随后用作在参考基因组中寻找精确匹配的种子。
- 然而,长读段映射的一个挑战是从每个长读段中提取的大量短种子。
- 为了应对这个问题并实现快速映射速度,通常使用一种基于最小代表性种子集的新方法。
- 这种最小化器方法提供了一种快速采样和总结长序列中k-mer的方法,基于此可以更容易地评估两个长序列的相似性。
- 基于这种方法,如果两个长序列共享足够长度的相同子序列,那么将为这两个子序列选择相同的k-mer(或最小化器)。
- 通过采用最小化器,定位两个长序列中共享的子序列在计算上变得更加可行。
- 以minimap2为例,索引参考基因组的步骤是通过在哈希表中索引参考基因组的最小化器来实现的。
- 为了映射长读段,查询读段的最小化器用作种子,以在参考基因组中找到精确匹配。
- 然后,将共定位的种子链接在一起作为扩展步骤的链。
para
- 除了映射算法外,当存在多个参考基因组序列时,参考基因组序列的选择也会影响映射结果。
- 根据大多数当前映射器的设计,与所选参考序列更相似的读取比那些与参考序列差异较大的读取对齐得更好。
- 如果差异足够大,它可能会被视为不匹配而被丢弃。
- 因此,使用不同的参考基因组序列可能会引入"参考偏差"。
- 使用任何一个特定的参考基因组不可避免地会引入这种偏差,因为单个参考基因组根本无法容纳在种群或物种中自然存在的序列变异和多态性。
- 应牢记这种偏差,尤其是当源生物的遗传背景与参考基因组不同时。
- 在这种情况下,比较使用不同参考基因组得到的映射结果可以帮助选择一个更合适的参考基因组。
- 为了对抗参考偏差,更近期的映射策略包括使用"主要等位基因"参考基因组,其中每个变异位点使用最常见的等位基因;多个参考基因组,这是参考流方法所使用的;或基于图的参考基因组(参见第16章,第16.6节)。
5.3.3 SAM/BAM as the Standard Mapping File Format
5.3.3 SAM/BAM 作为标准映射文件格式
para
- 由各种算法生成的映射结果通常存储在SAM或BAM文件格式中。
- SAM代表序列对齐/映射,具有制表符分隔的文本格式。
- 它可人工阅读且易于检查,但解析速度相对较慢。
- BAM是SAM的压缩二进制版本,体积更小且解析速度更快。
- 由于它们的广泛使用,SAM/BAM已成为存储读取映射结果的实际标准。
- SAM/BAM文件的基本结构简单明了,包含一个头部区域(可选)和一个对齐区域。
- 如果存在头部区域,它提供有关SAM/BAM文件的一般信息,并位于对齐区域之上。
- 头部区域中的每一行都以符号"@"开头。
- 对于对齐区域,有11个必填字段(列于表5.2中)。
- SAM/BAM格式的示例展示在图5.7中。
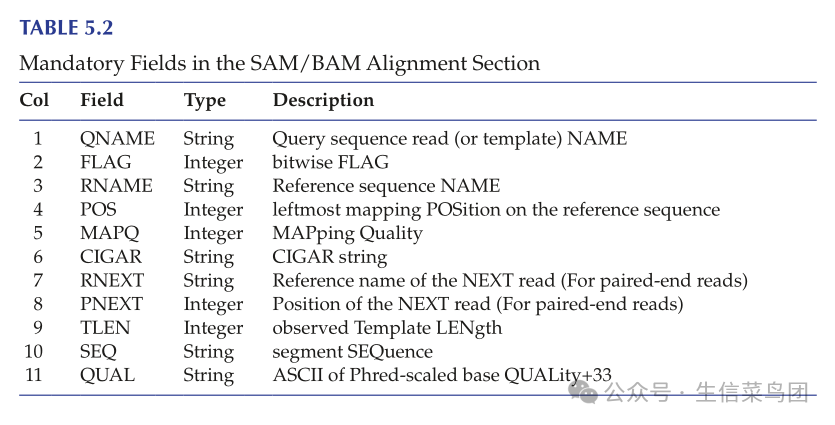
image-20241028015223958
para
- 在图5.7所示的示例中,头部部分包含两行。第一行有两个字母的记录类型代码HD,表示它是头部行,如果存在的话,它总是第一行。
- 这个记录有两个标签:VN,表示格式版本,和SO,表示排序顺序(在这种情况下,对齐是按坐标排序的)。
- 第二行是SQ,即参考序列字典。它也有两个标签SN和LN,分别表示参考序列名称和参考序列长度。
- 对于对齐部分,尽管表5.1中列出的多数字段是自解释的,但有些字段可能在一开始并不那么清楚。
- FLAG字段使用一个简单的十进制数字来跟踪映射过程中使用的11个标志的状态,例如是否存在多个测序段(如示例中的r001)或SEQ是否为反向互补。
- 要检查这些标志的状态和含义,需要将十进制数字转换为二进制形式。
- 对于POS字段,SAM使用1基的坐标系统,即参考序列的第一个碱基计为1(而不是0)。
- MAPQ是映射质量分数,其计算方式类似于之前介绍的Q分数(MAPQ = −10 × log10(PMapErr))。
- CIGAR(或简明特异间隙对齐报告)字段详细描述了SEQ如何映射到参考序列,包括标记SEQ中存在而参考序列中不存在的额外碱基,或SEQ中缺失的参考碱基。
- 在上面的示例中,r001/1的CIGAR字段显示值为"8M2I4M1D3M",这意味着前八个碱基与参考序列匹配,接下来的两个碱基为插入,接下来的四个与参考序列匹配,接下来的一个为删除,最后三个再次与参考序列匹配。
- 更多详情(例如不同FLAG状态)和SAM/BAM格式的完整规范,请参考SAM/BAM格式规范工作组的文档。
- 需要注意的是,BAM也可以用于存储未对齐的原始读取,作为该格式的一种"非标签"用途。
- 例如,PacBio平台以未对齐的BAM格式输出原始测序读取。
- 除了SAM/BAM格式外,另一种存储对齐读取的格式是欧洲生物信息学研究所设计的CRAM。
- CRAM文件比等效的BAM文件小,因为CRAM使用基于参考的压缩方案,即只存储与参考序列不同的碱基。

image-20241028015304388
- 图 5.7 存储NGS读段对齐结果的SAM/BAM格式。面板(a):显示与顶部显示的参考序列及其相应坐标的对齐。从参考序列派生的序列包括成对读段r001/1和r001/2。r002中的小写碱基表示与参考序列的不匹配,并在对齐过程中被剪裁。读段r003代表一个剪接对齐。面板(b):展示了SAM格式,其中包含11个强制字段,这些字段在表5.2中有详细说明。
5.3.4 Mapping File Examination and Operation
5.3.4 映射文件检查与操作
para
- 在执行映射过程后,应仔细检查SAM/BAM文件中报告的映射结果。
- 首先,应生成总结统计信息,例如对齐读数的百分比,特别是唯一映射读数的百分比。
- 目前,映射率仍然远低于100%。
- 即使在理想条件下,大多数对齐工具也只能为70-75%的序列读数找到唯一的基因组位置匹配。
- 这种无法确定大量读数的基因组起源的情况可以归因于多种因素,包括大多数基因组中存在的重复序列、大多数短NGS读数的相对较短长度以及因此有限的定位信息、算法限制、测序错误,以及种群中的DNA序列变异和多态性。
- 随着读数长度的增加和该领域积极发展中更好的算法设计,映射性能会得到改善。
para
- 其次,映射到多个基因组位置的读取,通常称为多读(multireads),通常不会对后续分析有所贡献,因此应该被过滤掉。
- 多读映射的模糊性是由于上述由多态性和突变引起的序列偏差、测序错误以及基因组中存在高度相似的序列,例如来自重复基因的序列。
- 将这些读取包含在下游分析中可能导致偏倚或错误的结果。
- 对于大多数实验,这些读取应该被排除在进一步分析之外。
- 由于过滤多读通常会移除大量读取,这可能导致潜在的信息丢失,有一些算法(如BM-Map)旨在通过概率分配将多读重新分配到竞争的基因组位点。
para
- 第三,除了多重读取外,重复读取也应在许多实验中被识别并过滤掉。
- 在一个多样化的非富集测序文库中,由于片段化过程的随机性,获得相同片段的几率极低。
- 即使通过PCR步骤来富集DNA片段,生成重复读取的几率仍然非常低(通常<5%),因为PCR过程中的循环次数有限,且后续的测序过程是对DNA文库的随机抽样(深度不一)。
- 因此,存在大量重复读取表明PCR过度扩增。
- 重复读取可以根据序列同一性来检测,但由于测序错误,这往往会低估重复读取的数量。
- 因此,更合适的方法是在映射步骤后检测重复读取(图5.8)。
- 由于PCR过度扩增导致的技术重复和真正的生物重复是不可区分的,研究者在决定是否从进一步分析中去除重复读取时应谨慎。
- 虽然去除重复读取在许多情况下(如变异发现)可以提高后续分析的性能,但在涉及较少复杂或 mostly enriched 的测序目标的情况下,包括那些来自极小基因组的,或用于RNA-seq或ChIP-seq的,去除它们可能导致丢失真正的生物信息。
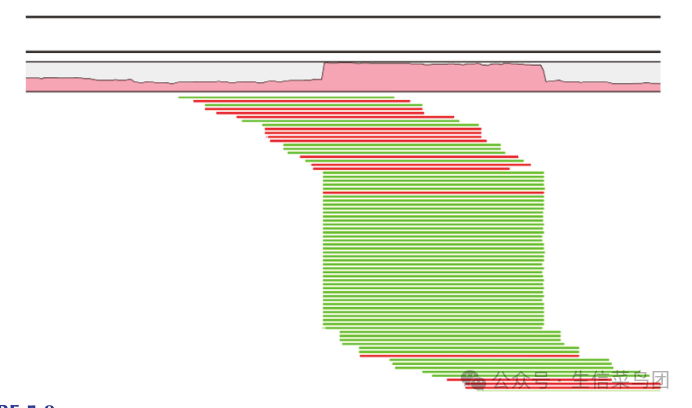
image-20241028015442730
- 图5.8 映射过程后重复读取的检测。顶部显示了参考基因组区域的覆盖深度。下方展示了映射读取以及一组映射到同一区域的重复读取。绿色和红色表示两条DNA链。(由CLC Genomics Workbench生成,并经CLC Bio许可使用。)
para
- 此外,还可以进行多种其他步骤来操作SAM/BAM文件。
- 这些步骤通常由SAMtools和Picard提供,这两个是广泛用于操作SAM/BAM文件的软件包。
- 这些操作包括:
para
- SAM和BAM的相互转换。SAMtools还可以将其他对齐文件格式转换为SAM/BAM
para
- 将多个BAM文件合并为单个BAM文件
para
- SAM/BAM文件的快速随机访问索引
para
- 使用各种标准对读取的比对进行排序,例如基因组坐标、泳道、文库或样本
para
- 额外的读段对齐过滤,例如移除只有一对中的一个映射到参考基因组的成对读段
para
- 生成一个 pileup 格式的文件(图 5.9),以显示每个基因组坐标上来自不同读段的匹配(或不匹配)碱基(SAMtools)
para
- 使用基于文本的查看器进行简单可视化,以便在较小的基因组区域内仔细检查读数对齐(SAMtools)

image-20241028015538037
- 图5.9 由SAMtools生成的pileup文件格式。pileup文件显示了映射读取中的测序碱基如何与每个基因组坐标的参考序列对齐。列包括(从左到右):染色体(或参考名称)、基因组坐标(从1开始)、参考碱基、映射到该碱基位置的总读取数、读取碱基及其调用质量。在读取碱基列中,点表示与参考碱基匹配,逗号表示与互补链匹配,‘AGCT’表示不匹配。此外,‘$’符号标记读取的结束,而‘^’标记读取的开始,‘^’后的字符表示映射质量。
para
- SAMtools 和 Picard 在处理和分析 SAM/BAM 文件方面非常灵活。
- 事实上,前面提到的步骤,即生成比对摘要统计和去除多重读数和重复读数,可以直接使用这些工具进行。
- 例如,SAMtools 和 Picard 都有用于检测和去除重复读数的工具,分别称为 markdup 和 markduplicates。
- 这些工具会将映射到相同起始基因组位置的读数标记为重复读数。
markdup markduplicates
para
- 最后,在检查映射结果方面,没有什么能替代在参考基因组背景下直接可视化映射读段的方法。
- 虽然基于文本的比对查看器,如SAMtools提供的工具,提供了一种简单的方法来检查小范围的基因组区域,但通过将映射读段序列覆盖在参考基因组上进行直接图形可视化,提供了更直观的数据检查和模式查找方式。
- 这种可视化过程服务于多种目的,包括额外的数据质量控制、实验程序验证和映射模式识别。
- 常用的可视化工具包括综合基因组查看器(IGV)、Artemis、SeqMonk、JBrowse和Tablet。
- UCSC和Ensembl基因组浏览器也通过添加自定义的BAM轨道提供了可视化选项。
- 映射后的数据质量控制工具,如Qualimap 2,也提供了关于关键指标的可视化总结,包括参考基因组的整体覆盖情况。
5.4 Tertiary Analysis
5.4 三级分析
para
- 序列读段映射步骤之后,后续分析因应用而异。
- 例如,RNA-seq数据分析的工作流程与突变和变异发现的工作流程不同。
- 因此,在本章中不可能提供一个适用于所有NGS数据分析的"典型"工作流程,除了数据质量控制、预处理和读段映射这些常见步骤。
- 第三部分中的章节提供了关于应用特定的三级分析步骤和常用工具的详细信息。
6 Computing Needs for Next-Generation Sequencing (NGS) Data Management and Analysis
para
- 我们生成下一代测序(NGS)数据的能力与我们从这些数据中提取知识的能力之间的差距正在扩大。
- 为了管理和处理海量的NGS数据,以深入了解生物系统,需要在计算基础设施和分析能力上进行大量投资。
- 然而,如何评估计算需求并构建一个满足这些需求的系统,对小型研究小组甚至大型研究机构都构成了严峻的挑战。
- 为了应对这一前所未有的挑战,NGS领域可以借鉴其他"大数据"领域的解决方案,如高能粒子物理学、气候学和社会媒体。
- 对于没有太多生物信息学训练的生物学家来说,虽然需要获得专家的帮助,但对NGS数据管理和分析的各个方面有一个良好的理解,对未来多年都是有益的。
6.1 NGS Data Storage, Transfer, and Sharing
6.1 NGS数据存储、传输与共享
para
- NGS已经成为科学研究大数据的主要生产者。
- 随着测序成本的不断下降,NGS数据产生的速度只会加快。
- 这意味着对更多数据存储、访问和处理能力的需求也随之增加。
- 与其他生物检测生成的文件相比,如凝胶图片甚至微阵列数据文件,NGS文件要大得多。
- 对于一个个体实验室来说,单次典型运行生成的数据在压缩FASTQ格式下可达数十到数千千兆字节(GB)。
- 在与参考基因组对齐后,处理过的文件大小显著增加。
- 进一步的分析导致更多文件的生成和数据量的扩散。
- 为了容纳多次运行的原始和处理过的文件,需要数十太字节(TB)或更多的存储空间。
- 存储和归档这些文件绝非易事。
- 更糟糕的是,单次运行的原始测序信号强度文件,如扫描图像或视频,规模可达TB级别(这一数量未计入上述数据量中)。
- 随着这些原始信号文件的积累,它们很容易压垮大多数数据存储系统。
- 虽然这些原始图像文件可以长期保留,但较新的测序系统会在分析过程中实时处理它们,并在默认情况下一旦分析完毕即删除,以减轻存储负担。
- 通常情况下,如果数据丢失,重新运行样本比归档这些巨大的原始信号文件更容易且更经济。
para
- 由于大多数NGS文件体积巨大,从一个地方传输到另一个地方并非易事。
- 对于一个小型项目,将测序文件从生产服务器传输到本地存储空间,如果网络连接速度快,通过FTP或HTTP下载可能足够。
- 至于网络速度,1 Gbps的网络是必要的,而10/100 Gbps的网络在高流量情况下能提供更好的性能。
- 当网络速度较慢或需要传输的数据量过大时,使用外部硬盘可能是唯一的选择。
- 当数据到达实验室后,为了快速进行本地文件读取、写入和处理,它们需要存储在专用工作站或服务器内的硬盘阵列中。
para
- 对于生产环境,例如NGS核心设施或大型基因组中心,这些机构为大量项目生成NGS数据,需要企业级数据存储系统,如DAS(直接附加存储)、SAN(存储区域网络)或NAS(网络附加存储),以提供具有高可靠性、访问速度和安全性的集中式数据存储库。
- 为避免意外数据丢失,这些数据存储系统通常会被备份、镜像或同步到分布在不同位置的数据服务器上。
- 对于涉及多个站点和数百PB至EB级数据的大规模协作项目,数据传输和共享的过程带来了更多挑战,这促使了如Globus等高容量和高性能平台的发展。
para
- 协作组之间的数据共享会带来超出单个实验室处理范围的技术问题。
- 相比于在多个地点简单复制数据,一个集中的数据存储库可能更受欢迎,以促进有效协作和及时讨论。
- 随着数据共享而来的还有数据访问控制和来自以患者为导向的研究数据的隐私问题。
- 从更广泛的意义上讲,与整个生命科学社区共享下一代测序(NGS)数据也增加了研究项目的价值。
- 因此,许多期刊实施数据共享政策,要求在发表前将序列读取数据和处理后的数据存入公开可访问的数据库(例如NCBI的序列读取存档[SRA]或欧洲核苷酸档案[ENA])。
- 为了便于数据解读和潜在的荟萃分析,与该实验相关的信息也必须与数据一同存档。
- 一些组织,如功能基因组数据学会,已经制定了关于应与数据一同存档的信息的指南。
- 例如,MINSEQE(高通量核苷酸测序实验最低信息)指南指定了以下信息应与序列读取数据和处理后的数据一同提供:生物系统、样本和实验变量的描述;实验概要和样本数据关系;基本的实验和数据处理协议。
para
- 为社区归档下一代测序数据及其相关信息是一项庞大的任务,需要大量投资来维护和发展必要的基础设施和专家支持。
- 美国国家生物技术信息中心(NCBI)的序列读取档案(SRA)存储库在2011年因高昂成本和政府预算限制而关闭。
- 然而,由于其对社区至关重要,美国国立卫生研究院(NIH)在同年底恢复了对其的支持。
6.2 Computing Power Required for NGS Data Analysis
6.2 NGS数据分析所需的计算能力
para
- 处理大量下一代测序(NGS)数据需要大量的计算能力。
- 需要多少计算能力的问题取决于要进行的分析类型。
- 例如,对大型基因组的从头组装需要比变异发现的重新测序或用于鉴定差异表达基因的转录组分析多得多的计算能力。
- 因此,为了确定一个项目、实验室或组织所需的计算能力,首先需要分析将要进行的NGS工作类型。
- 如果工作需要密集计算,或者涉及新算法和软件工具的开发和优化,可能需要一个高性能集群。
- 另一方面,如果工作将使用不需要高度密集计算的成熟工作流程,一个强大的工作站可能就足够了。
- 还建议计算机系统可扩展,以适应未来计算需求的增加,这是由于未来研究项目不可预见的变化或高通量基因组学技术的进一步发展。
para
- 对于一个小型项目,进行NGS数据分析所需的最基本系统可以简单为一个64位计算机,配备8 GB的RAM和两个2-GHz的四核处理器。
- 使用这样的计算机,可以对获得的序列读数进行基本的参考基因组映射。
- 这种基本设置允许一次处理一个数据集。
- 为了同时处理多个数据集或项目,需要具有更多内存和CPU核心的高性能计算(HPC)系统。
- HPC系统所需的核心数量取决于一次需要同时运行的任务数量。
- 对于每个任务,所需的核心数量依赖于任务的性质和执行该任务的算法。
para
- 除了CPU核心数量外,系统的内存量也极大地影响其性能。
- 同样,内存需求取决于要处理的作业数量和复杂性,例如,将读取映射到小型基因组可能只需要几GB的内存,而大型基因组的从头组装可能需要数百GB甚至TB级别的内存。
- 目前的估计是,每个CPU核心所需的内存量不应少于3GB。
- 在早期使用SOAPdenovo管道进行人类基因组从头组装的实现中(将在第12章详细说明),使用了配备32个核心(八个AMD四核2.3 GHz CPU)和512 GB内存的标准超级计算机。
- 作为近期从头基因组组装所需计算能力的一个例子,一个瑞典团队使用了一台配备64个核心(八个Intel Xeon X6550 8核2.00 GHz CPU)和2 TB RAM的服务器。
- 对于小型基因组(如微生物基因组)的从头组装,一台至少包含8个CPU核心、256 GB RAM和快速数据存储系统的机器可以在合理的时间框架内完成工作。
- 根据目前的估计,一台配备8个核心、32 GB RAM和10 TB存储的工作站可以满足许多不进行从头基因组组装的项目需求。
para
- 完成一项工作所需的时间因工作的复杂性和可用的计算能力而大不相同。
- 以更具体的例子来说,运行基于深度学习的全基因组测序变异调用工具DeepVariant(详见第10章)在使用最低配置的8核计算机和16 GB内存时需要24至48小时,但使用带有4 GB专用视频内存和CUDA并行计算支持的图形处理单元(GPU)时,处理时间减少了一半以上。
- 使用Bowtie在配备32核和128 GB内存的计算机上将8000万个75-bp读段映射到人类基因组,耗时不到2小时,而在后续步骤包括标准化和差异表达统计测试中所需时间更少。
- 在一项小型RNA-seq研究中,使用32核和132 GB内存的工作站处理20个多重条形码样本,总共有1.6亿个读段,样本去复用耗时稍超过2小时,读段映射到宿主基因组和小RNA注释数据库的时间也大致相同。
6.3 Cloud Computing
6.3 云计算
para
- 如上所述,NGS数据存储、传输和共享绝非易事。
- 本地构建的计算系统的一个局限性是其可扩展性。
- 随着NGS技术进步和测序成本下降的速度快于计算机硬件行业的发展速度,NGS数据生成与我们处理和分析它们的能力之间的差距只会越来越大。
- 为了缩小这一差距并加快NGS数据处理速度,NGS社区已经从长期存在的本地计算模式转向云计算模式。
- 诸如亚马逊、微软和谷歌等公司一直在构建超大规模的云计算集群和数据存储系统,供终端用户通过互联网使用。
- 与本地计算相比,云计算使得用户能够访问超级计算和大规模数据存储能力,而无需构建和维护本地工作站、服务器或HPC集群。
para
- 云计算的核心是虚拟化技术,它允许终端用户按需创建虚拟计算机系统,并灵活指定所需的CPU核心数量、内存大小、磁盘空间以及操作系统。
- 通过这项技术,多个虚拟计算机系统可以同时在同一台物理云服务器上运行。
- 将云计算应用于下一代测序(NGS)数据处理已经展示了这种"按需超级计算"模型的优势,包括灵活性、可扩展性以及通常情况下的成本节约。
- 云计算提供的灵活性和可扩展性使研究人员能够利用以前仅存在于大型基因组中心的超级计算能力进行NGS数据分析。
- 成本节约的实现是因为用户只需为用户配置的计算实例使用的时间付费。
para
- 使用云的另一个优势在于研究人员和项目之间的数据共享。
- 通过提供单一的集中式数据存储,云使得位于不同地理位置的不同团队能够访问相同的数据集并共享分析结果。
- 此外,借助云计算,将软件工具应用于"大型"NGS数据的任务可以更容易实现。
- 与NGS数据文件的大尺寸相比,设计用于处理它们的软件和脚本要小得多。
- 因此,下载和安装它们到数据存储的位置要容易得多,也更为高效,而不是将大量NGS数据移动或复制到工具安装的位置。
- 通过直接在云中存储生产数据,数据传输的负担大大减轻;通过将数据和工具耦合在同一位置,可以实现最佳性能。

image-20241028020036983
para
- 尽管云计算使用户能够减轻运行和维护本地计算系统的麻烦和成本,但它确实存在一些需要考虑的缺点。
- 迁移到云的一个实际障碍是数据传输到云中和从云中传输的速度。
- 使用低速互联网连接将100 GB的数据上传到云可能需要一周时间。
- 是否在云中运行分析在很大程度上取决于要传输的数据量以及分析步骤的计算复杂性。
- 通常情况下,只有当分析任务每字节数据需要超过10^5个CPU周期时,才值得将数据上传到云进行处理。
- 因此,对于处理大量数据但不涉及大量高计算强度步骤的项目,花费在数据传输到云上的时间可能会比数据处理的时间更多。
- 其他潜在因素包括数据安全性、在某些情况下成本效益不高、云环境中分析工具的可用性以及网络停机时间。
- 虽然用户可以从互联网上的任何地方访问他们的数据,但这种便利也意味着数据安全可能被破坏或泄露的可能性。
- 一些重度用户可能会发现云计算不如运行本地服务器划算。
- 尽管越来越多的工具在云中变得可用,用户仍需谨慎确保他们所需的工具是可用的。
- 对于那些经常遭受网络中断的地方的用户来说,云计算可能会出现问题,因为所有基于云的操作都依赖于互联网流量。
para
- 尽管存在潜在的不足,云计算已被证明是下一代测序(NGS)数据分析的一种可行方法。
- 表6.1列出了当前可用于NGS应用的一些云计算提供商。
- 为了说明如何利用云计算进行NGS数据分析,下面以使用亚马逊弹性计算云(EC2)进行读段对齐为例。
- 首先,将输入数据文件(FASTQ文件和参考基因组文件)从本地计算机上传到亚马逊简单存储服务(S3)中的"桶"中。
- 这个云存储桶也用于存放程序脚本和输出文件,可以通过亚马逊网络服务(AWS)管理控制台创建,这是一个访问所有亚马逊云资源的统一界面。
- 为了启动对齐,首先必须使用控制台的"创建工作流"功能定义工作流。
- 定义工作流时,需要指定输入序列读段文件、对齐器脚本以及对齐输出文件的保存位置。
- 同时,还需要配置完成该任务所需的亚马逊EC2实例数量,这将决定内存和处理器分配。
- 配置完成后,通过管理控制台提交任务。
- 当实例完成时,对齐输出文件将被存放到S3云存储中预先指定的文件位置。
6.4 Software Needs for NGS Data Analysis
6.4 NGS数据分析所需的软件
para
- 要配置云实例或设置本地工作站或服务器,需要选择和安装操作系统和软件。
- 虽然一些NGS分析软件(如CLC Genomics Workbench)可以在Windows环境中运行,但大多数工具只能在Unix(或Linux)环境中运行。
- 因此,Unix或Linux通常是安装在这样一台机器上的操作系统。
- 在Unix或Linux中安装软件并不像在Windows中那样简单,因为从开发者网站下载的未编译的软件源代码需要先进行编译,然后才能安装到特定版本的操作系统上。
- 如果读者不熟悉Unix/Linux环境及其使用的命令行界面,建议阅读入门书籍或基于网络的教程。
para
- 一种减少使用为Unix/Linux环境开发的工具障碍的方法是通过一个"桥接"系统访问它们,例如Galaxy,它为命令行工具提供了一个更友好的用户界面。
- 由宾夕法尼亚州立大学的Nekrutenko实验室和约翰霍普金斯大学的Taylor实验室开发的Galaxy系统,提供了一种通过熟悉的网页浏览器界面部署这些工具的机制,使它们能够被不同操作系统的用户访问。
- Galaxy系统具有高度的可扩展性,最新的工具不断被封装以便通过网页界面执行。
- 除了提供用户友好的界面外,这样的系统还允许从不同的工具创建数据分析工作流程,这使得能够快速部署多个工具协同工作,实现一致性和可重复性,并与其他研究人员共享分析过程。
- Galaxy可以通过公共服务器(例如usegalaxy.org)访问,安装在本地实例上,或在云端使用。
- 使用公共服务器时,用户无需维护本地服务器,但分配给每个账户的可用存储空间通常有限,且计算资源与其他许多用户共享。
- 在Unix/Linux或Mac OS上创建本地Galaxy实例需要一些努力,用户确实需要提供维护,但用户对存储空间、计算能力和从Galaxy工具库(目前接近10,000个工具)中选择和安装工具有更多控制权。
- Galaxy团队通过提供详细且易于遵循的说明,使得安装本地实例变得非常容易。
- Galaxy也可在云端通过AnVIL中的专用实例使用,AnVIL是一个基于谷歌云平台(GCP)并由美国国家人类基因组研究所支持的数据分析、存储和管理平台。
- 除了提供高度可扩展的计算环境外,AnVIL中的Galaxy云实例还提供对私有数据集的保护,同时促进协作。
para
- 还有一些其他社区项目提供了替代平台,以方便用户访问各种NGS和其他基因组分析工具。
- Bioconductor是一个开源和开放开发的软件项目,是这些项目中最为知名的。
- 这个大规模项目基于R语言,这是一种专为统计计算和图形设计的编程语言和软件环境。
- 旨在为高通量基因组数据的分析和理解提供工具,Bioconductor软件库在撰写本文时包含超过2200个软件包,其中许多是设计用来处理NGS数据或可用于处理NGS数据的。
- R环境和Bioconductor库可以安装在所有主要操作系统上,包括Windows。
- Bioconductor项目门户网站(www.bioconductor.org)和R项目网站(www.r-project.org)提供了这些软件包的安装和使用详细信息和教程。
- 每个工具都有详尽的文档,并提供了实际使用示例。
para
- 识别、安装和维护适用于本地Unix/Linux工作站、本地Galaxy实例或本地Bioconductor R库的合适NGS分析软件,并不是一件简单的事情。
- 新的软件工具不断被开发和引入,而许多现有的工具也时常更新。
- 为了评估候选软件包并识别适合安装和使用的工具,最好使用多个测试数据集,不仅包括计算机模拟的数据,还包括来自真实世界生物样本的数据。
- 此外,几乎所有的工具都有可调整的参数,这些参数应设置得一致,以便于性能比较。
- 在性能方面,早期的NGS软件通常没有利用高性能并行计算的优势。
- 为了提高性能并充分利用高性能计算系统中的多个核心或节点,较新的算法倾向于使用线程或多进程通信接口(MPI)来将工作分散到多个进程中。
- 因此,在评估NGS工具时,检查是否采用了这些类型的并行处理以利用多核计算架构的优势也是很有帮助的。
6.4.1 Parallel Computing
6.4.1 并行计算
para
- 并行化是一个计算术语,描述将任务分解为多个独立子任务的过程,可以显著提高高度可并行任务的处理速度,其中包含许多NGS数据分析步骤。
- 例如,尽管一次测序运行会生成数百万个读数,但这些读数映射到参考基因组的过程是"尴尬并行"的,因为每个读数都是独立映射到参考基因组的。
- 由于并行计算可以由GPU高效执行,因为计算机屏幕上每个像素的渲染也是一个高度并行的过程,将GPU与CPU集成在异构计算系统中可以增加10到100倍的吞吐量,并将单个计算机转变为迷你超级计算机。
- 虽然这些系统可以应用于NGS数据分析的各个方面,但许多NGS分析工具尚未充分利用此类系统中并行计算的力量。
para
- 并行化也是决定 CPU(或 GPU)核心数量增加如何影响实际 NGS 数据处理性能的重要因素。
- 如果一个步骤高度可并行化,并且为其设计的算法采用了并行化,那么核心数量的增加很可能会提高性能。
- 相反,如果该步骤不易并行化,或者即使任务可并行化但所部署的算法未使用并行化,仅仅拥有更多核心可能并不会提升性能。
6.5 Bioinformatics Skills Required for NGS Data Analysis
6.5 用于NGS数据分析所需的生物信息学技能
para
- 对于生物学家和生命科学领域的学生来说,掌握基本的生物信息学技能是非常有利的,因为生物学已经变得越来越数据丰富和数据驱动。
- 了解生物信息学的基础知识也有助于与生物信息学家进行交流,以便进行更高级的任务。
- 一般来说,这些技能包括使用常见的计算环境、生物信息学算法和软件包。
- 以下是生物学家处理NGS数据所需的生物信息学技能的简短列表:
para
- 熟悉Unix/Linux,以及Unix/Linux计算环境中最常用的命令。这对于在HPC集群上运行作业、在运行Unix/Linux操作系统(如Ubuntu或macOS)的本地机器上,或者在提供兼容层以在Windows上原生运行Linux工具的Windows子系统(WSL)中运行作业是必不可少的。
para
- 用于NGS数据分析的常用编程语言的基础知识。
- 这些语言包括R、Perl和Python,它们都是开源的,易于学习,并且拥有庞大的用户基础以提供帮助和支持。
- 虽然编程对生物学家来说不是必需的,但了解算法如何逐步执行是有帮助的,尤其是当现有的工具在特殊情况下无法理想工作时,需要进行修改。
para
- 了解计算生物学和生物统计学中的关键概念。
- 一些在计算机科学领域,特别是机器学习和数据挖掘中发展的计算方法,已被广泛应用于高通量生物数据处理。
- 人工神经网络(ANNs)、隐马尔可夫模型(HMMs)和支持向量机(SVMs)在此领域内是很好的例子。
- 诸如线性回归和非线性回归的统计方法被整合到许多基因组数据分析工具中,也应被纳入生物学家知识库中。
para
- 对关系型数据库的基本理解。目前用于注释和解释NGS数据的绝大多数信息都被捕获在各种数据库中。
- 了解数据库设计和结构是提取、操作和处理这些数据库中存储的信息以生成新的生物学知识的基础。
- 知道如何通过SQL(标准查询语言)或APIs(应用程序编程接口)与数据库交互也是有益的。
- 对关系型数据库及其操作的了解也有助于生物学家整理、组织和传播从NGS项目中产生的海量信息。
para
- 对计算机硬件如CPU、RAM和存储的基本理解和处理。
- 尽管严格来说计算机硬件不属于生物信息学的范畴,但了解如何组装工作站并投入使用仍然是有利且经济的。
- 了解高性能计算集群或异构计算系统通过并行处理的工作原理也是有益的,因为设计用于利用这些计算系统的NGS工具通常性能更好,这种知识可以帮助评估和选择那些在计算系统中最大化性能的工具。
para
- 对于处理下一代测序(NGS)数据的生物信息学家来说,以下是一些所需的技能列表:熟练使用基于Unix的操作系统;
- 熟悉一种编程语言,如Python、Perl、Java或Ruby;
- 熟悉统计软件,如R、MATLAB或Mathematica;
- 了解超级计算、高性能计算(包括并行计算)和企业数据存储系统;
- 掌握数据库管理语言,如MySQL或Oracle;
- 熟悉网页制作和基于网络的用户界面实现技术;
- 了解分子生物学与遗传学、细胞生物学和生物化学。
腾讯云开发者
