Python做单细胞测序数据分析必备技能
Python做单细胞测序数据分析必备技能

在前面单细胞转录组数据分析的文章中,有使用python实现的。今天我们了解一下在单细胞转录组分析过程中,必须要掌握的python数据框、包和函数等内容。
01、YEAR-END SUMMARY
数据结构
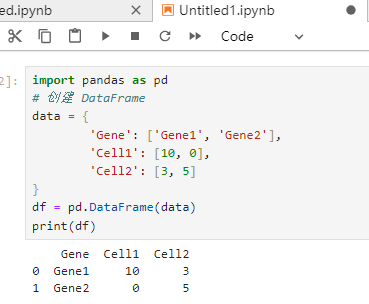
Pandas DataFrame
Pandas是Python中最常用的数据处理库,DataFrame是其核心数据结构,类似于Excel表格,便于数据的存储和操作。
import pandas as pd
# 创建 DataFrame
data = {
'Gene': ['Gene1', 'Gene2'],
'Cell1': [10, 0],
'Cell2': [3, 5]
}
df = pd.DataFrame(data)
print(df)
Numpy数组
NumPy提供了高效的多维数组对象,是数值计算的基础。
import numpy as np
# 创建 NumPy 数组
array = np.array([
[1, 2, 3],
[4, 5, 6]
])
print(array)
01、YEAR-END SUMMARY
「关键的Python包」
1. Pandas
用于数据加载、清洗和处理。常用函数包括read_csv(), merge(), groupby(), pivot_table()等。
# 读取数据
df = pd.read_csv('data.csv')
# 数据清洗
df.dropna(inplace=True)2. NumPy
用于高效的数值计算,常用于数组操作和数学运算。
# 数组运算
array = np.array([1, 2, 3])
array = array * 2
print(array)
3. Scanpy
专门用于单细胞分析的包,集成了数据预处理、降维、聚类和可视化等功能。
import scanpy as sc
# 读取数据
adata = sc.read_10x_h5('data.h5')
# 预处理
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.normalize_total(adata, target_sum=1e4)
4. Omicverse
Omicverse是一个集成的生物信息学工具包,支持多组学数据的分析。它提供了友好的接口,方便用户进行多种分析任务。
from omicverse import Omicverse
# 初始化 Omicverse 对象
ov = Omicverse(data='path_to_data')
# 执行分析
ov.run_analysis(method='scRNA-seq')
5. Matplotlib 和 Seaborn
用于数据可视化,帮助理解数据分布和分析结果。
import matplotlib.pyplot as plt
import seaborn as sns
# 绘制散点图
sns.scatterplot(
x='PC1',
y='PC2',
data=adata.obsm['X_pca']
)
plt.show()
YEAR-END SUMMARY
单细胞转录组分析流程中的关键函数
1.数据预处理
sc.pp.filter_cells(): 过滤低质量细胞。
sc.pp.filter_genes(): 过滤低表达基因。
sc.pp.normalize_total(): 数据归一化。
sc.pp.log1p(): 对数据进行对数转换。
2. 降维和聚类
sc.tl.pca(): 主成分分析(PCA)。
sc.pp.neighbors(): 构建邻近图。
sc.tl.leiden(): Leiden聚类算法。
sc.tl.umap(): UMAP降维。
3. 细胞注释
细胞注释是将聚类结果与已知细胞类型进行匹配的过程。常用的方法包括基于已知标记基因的注释和自动注释工具。
基于标记基因注释:
sc.tl.rank_genes_groups(): 识别各个簇的标记基因。
sc.pl.rank_genes_groups(): 可视化标记基因。
# 识别标记基因
sc.tl.rank_genes_groups(adata, 'leiden', method='t-test')
# 可视化标记基因
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)自动注释工具:
sc.tl.ingest()(需安装相关包):将已有的注释信息迁移到新的数据集。
scanorama、scmap 等工具可以用于自动注释。
4. 去批次效应
批次效应是不同实验批次之间的系统性偏差,去除批次效应有助于提高数据的整合性和分析的准确性。
sc.pp.combat(): 使用ComBat方法去除批次效应。
# 去除批次效应
sc.pp.combat(adata, key='batch')scanpy.external.pp.bbknn(): 使用BB KNN方法进行批次效应校正和邻居图构建。
import scanpy.external as sce
# 使用 BBKNN 进行批次效应校正
sce.pp.bbknn(adata, batch_key='batch')scanorama:
import scanorama
# 分割数据集
adatas = scanorama.utils.split_anndata(adata, 'batch')
# 整合数据
integrated, _ = scanorama.integrate_scanpy(adatas, dimred=50)
# 更新 adata
adata.obsm['X_scanorama'] = integrated.X 5. 差异表达分析
差异表达分析用于识别不同细胞群体之间显著差异表达的基因,这对于理解细胞功能和状态至关重要。
sc.tl.rank_genes_groups(): 识别差异表达基因。
# 差异表达分析
sc.tl.rank_genes_groups(adata, 'leiden', method='t-test')
# 查看结果
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)scanpy.tl.rank_genes_groups 支持多种方法,如't-test', 'logreg', 'wilcoxon'等。
6. 细胞组成分析
细胞组成分析用于评估不同条件或样本中各类细胞的比例变化,这对于理解生物学过程中的细胞动态非常重要。
sc.tl.louvain() / sc.tl.leiden(): 聚类细胞。
scanpy.external.tl.cell2location()(需安装相关包):空间定位和细胞组成分析。
sc.tl.score_genes(): 计算特定基因集的得分,用于评估细胞状态或类型。
proportion_analysis:使用自定义函数或第三方包进行细胞比例计算和比较。
import scanpy.external as sce
# 细胞组成分析
sce.tl.cell2location(adata, ...)
# 计算细胞周期得分
sc.tl.score_genes_cell_cycle(adata, s_genes, g2m_genes)
# 可视化细胞周期得分
sc.pl.scatter(adata, x='score_S', y='score_G2M')
# 计算每个簇的细胞比例
proportions = adata.obs['leiden'].value_counts(normalize=True)
print(proportions)7. 轨迹推断
轨迹推断用于推测细胞在发育或分化过程中的动态变化路径。
sc.tl.dpt(): 计算扩散潜在时间(Diffusion Pseudotime)。
scanpy.tl.paga(): 使用PAGA进行全局轨迹推断。
「示例全流程」
以下是一个包含上述关键函数的完整单细胞转录组分析示例,涵盖数据加载、预处理、降维、聚类、批次效应去除、细胞注释、差异表达分析和细胞组成分析等步骤。
import scanpy as sc
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
from omicverse import Omicverse
# 读取10X数据
adata = sc.read_10x_h5('data.h5')
# 预处理
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
# 识别高变基因
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
adata = adata[:, adata.var.highly_variable]
# PCA降维
sc.tl.pca(adata, svd_solver='arpack')
# 去除批次效应
sc.pp.combat(adata, key='batch')
# 邻近图和聚类
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
sc.tl.leiden(adata, resolution=0.5)
# UMAP可视化
sc.tl.umap(adata)
sc.pl.umap(adata, color=['leiden'])
plt.show()
# 细胞注释
# 识别标记基因
sc.tl.rank_genes_groups(adata, 'leiden', method='wilcoxon')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
# 差异表达分析
sc.tl.rank_genes_groups(adata, 'leiden', method='t-test')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
# 细胞组成分析
proportions = adata.obs['leiden'].value_counts(normalize=True)
print("细胞簇比例:\n", proportions)
# 可视化细胞组成
proportions.plot(kind='bar')
plt.ylabel('Proportion')
plt.title('Cell Cluster Proportions')
plt.show()
总结:
掌握Python在单细胞转录组分析中的应用,不仅能提高分析效率,还能深入理解数据背后的生物学意义。通过学习和实践,逐步熟悉Pandas、NumPy、Scanpy、Omicverse等关键工具,以及细胞注释、去批次效应、差异表达分析、细胞组成分析等关键函数,您将能够胜任从数据预处理到高级分析的各个环节。希望本文能为您的学习之路提供有益的指导!

腾讯云开发者
