高分杂志中的LASSO模型构建以及结果可视化一整套代码分析(Q1/IF: 58.7)

高分杂志中的LASSO模型构建以及结果可视化一整套代码分析(Q1/IF: 58.7)

生信技能树
发布于 2025-10-11 13:54:14
发布于 2025-10-11 13:54:14
前面我们给大家分享了一个将文章分析的数据和代码整理为在线书籍形式的文献,见:分享一篇将代码整理成在线book形式的顶刊文献(Q1/IF: 58.7),今天就来学习其中的Figure3,使用蛋白数据构建pdac预后模型。
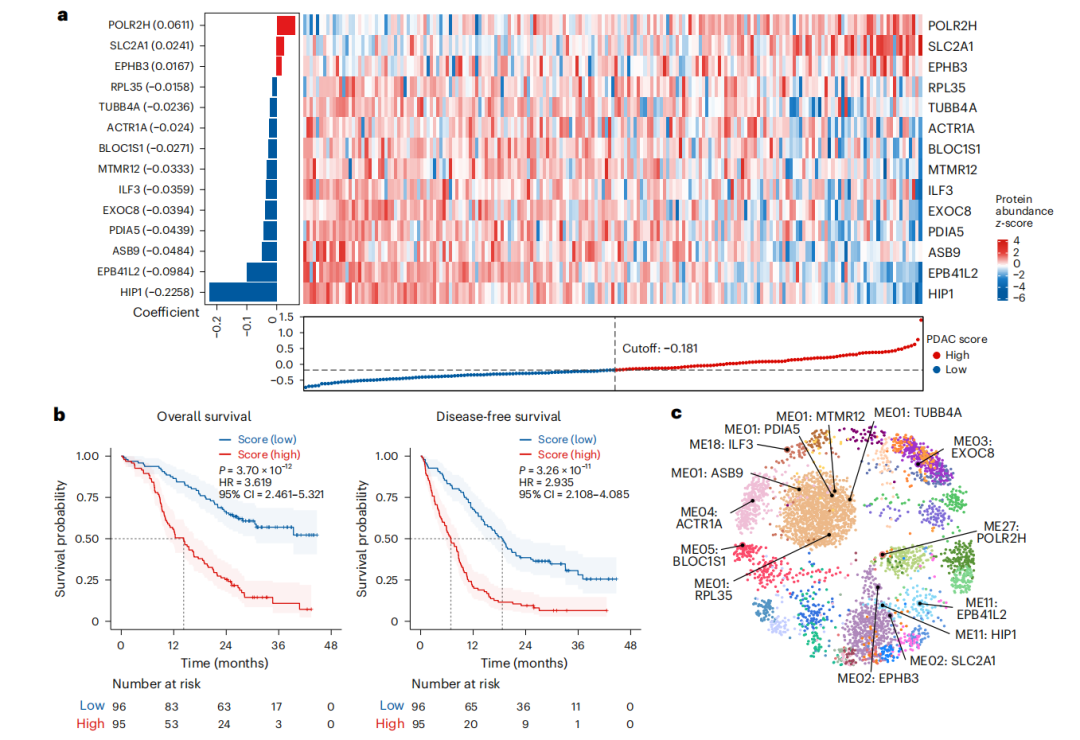
图三:建立最佳蛋白质组学水平的预后风险模型
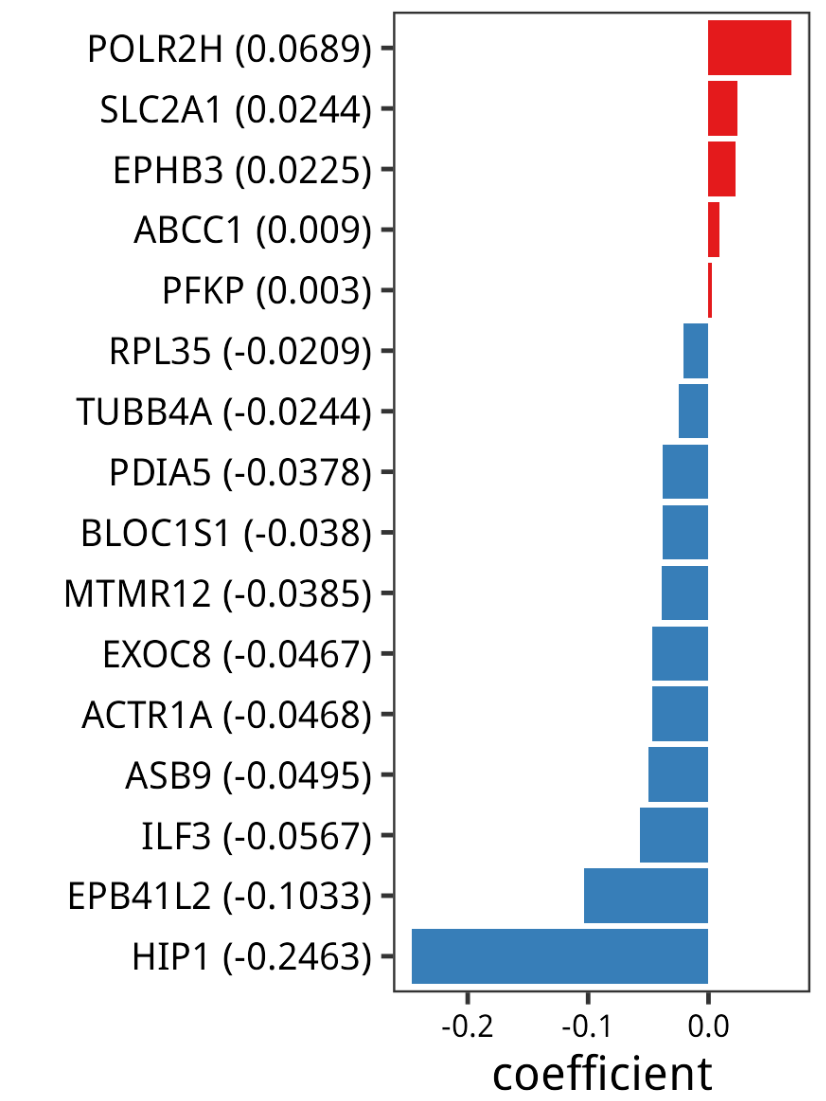
a,PDAC的LASSO模型。左侧面板显示LASSO-Cox回归模型中14种蛋白质的回归系数。
b,KM生存曲线比较“RJ队列1”中LASSO评分低和评分高组的OS(左侧)和DFS(右侧)。
c,将LASSO模型中的14种蛋白质叠加到加权相关网络节点上。节点按功能蛋白模块着色(与图2a相同),黑色节点代表LASSO模型中的蛋白质。
还有附图:
「代码和数据」
文献中所用的数据和代码分可以在下面两个地方下载得到:
http://www.genetictargets.com/PDAC2BOOKLET/index.html
构建PDAC的 LASSO model
通过最小绝对收缩和选择算子(LASSO)-Cox回归分析为胰腺导管腺癌(PDAC)建立了一个预后风险模型。该模型包含14种蛋白质(但是我跑出来的有16个蛋白,应该是这个 forestmodel 包的版本不一样导致的!),根据它们的LASSO评分将PDAC患者分为两组,即高分组和低分组。
step1: 加载数据
先读取蛋白表达矩阵,提取其中的肿瘤样本,然后再提取wgcna结果中的蛋白。
rm(list=ls())
library(glmnet)
library(openxlsx)
library(survminer)
library(survival)
library(tidyverse)
library(RColorBrewer)
library(ComplexHeatmap)
library(forestmodel)
library(ROCR)
dir.create("./results/Figure3/lasso",recursive = T)
# Step 1: load data
## PDAC Tumor proteomics
pdac.pro <- readRDS('./PDAC2_data_results/data/proteomics/20230412_PDAC_PRO_exp.rds')
dim(pdac.pro)
pdac.pro[1:5,1:5]
# 191 tumor samples * 4787 protein
pdac.pro <- pdac.pro [,grep('_T_',colnames(pdac.pro))] %>% t() %>% as.data.frame()
## WGCNA protein
wgcna <- readRDS("./PDAC2_data_results/data/proteomics/wgcna/2D-node.data.rds")
pdac.pro <- pdac.pro [,intersect(wgcna$Gene,colnames(pdac.pro))] # 191 samples * 3906 protein
dim(pdac.pro)
pdac.pro[1:5,1:5]
padc.pro 现在是一个191行样本,3906列蛋白的表达矩阵。
读取临床信息:
## PDAC clinical data
cli.pdac <- readRDS('./PDAC2_data_results/data/clinical/20230412_PDAC_191_patients_clinical.rds')
rownames(cli.pdac) <- cli.pdac$ID
head(cli.pdac)
step2:拟合 Lasso Regression 模型
使用表达矩阵 x.train,临床信息 y.train 来构建模型。
交叉验证:使用 cv.glmnet() 函数实现自动化交叉验证,选择最佳 λ(惩罚系数)。
# Step 2: Fit the Lasso Regression Model
## Lasso data
y.train <- data.frame(time=cli.pdac$Time_OS,
status=cli.pdac$Censoring_OS,
row.names = row.names(cli.pdac)) %>%
na.omit(y.train) %>%
as.matrix()
x.train <- pdac.pro[row.names(y.train),] %>% as.matrix() %>% na.omit()
y.train <- y.train[row.names(x.train),]
set.seed(1000)
fit <- glmnet(x=x.train,
y=y.train,
family = "cox", # cox : cox , binomial : logistic
alpha = 1) # 1 : lasso penalty, 0 : ridge penalty.
pdf('./results/Figure3/lasso/0.Coefficients.pdf',width = 5,height = 4)
plot(fit,xvar="lambda",label=F)
dev.off()
# Cross-validation
cvfit <- cv.glmnet(x=x.train,
y=y.train,
family = "cox",
type.measure="deviance",
alpha = 1,
nfolds = 10)
png(paste0('./results/Figure3/lasso/S5A-Coeff.png'), width = 5,height = 5,res=1200,units="in")
plot(fit,xvar="lambda",label=F)
abline(v = -log(cvfit$lambda.min), col = "black",lty=2)
text(4,4, labels = paste0("lambda.min: ", round(cvfit$lambda.min,4)),col = "black",cex = 0.75)
dev.off()
paste0("lambda.min: ", round(cvfit$lambda.min,4))
log(cvfit$lambda.min)
表达矩阵 x.train:
临床信息 y.train :
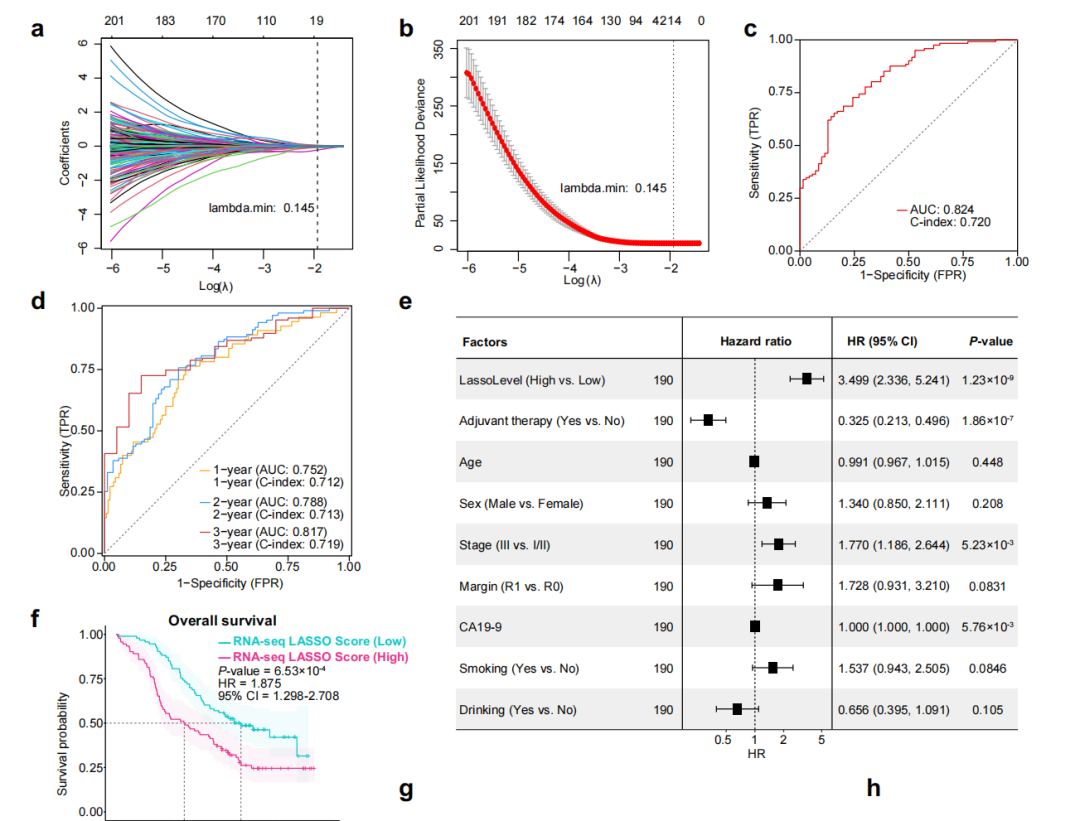
S5A-Coeff:LASSO 回归路径图:展示惩罚系数 λ 不同取值下各变量的回归系数变化。
注意这里的横坐标与原文有差别,这里有个负号,方向相反,但是没有啥问题。只需要从右向左看就好了。
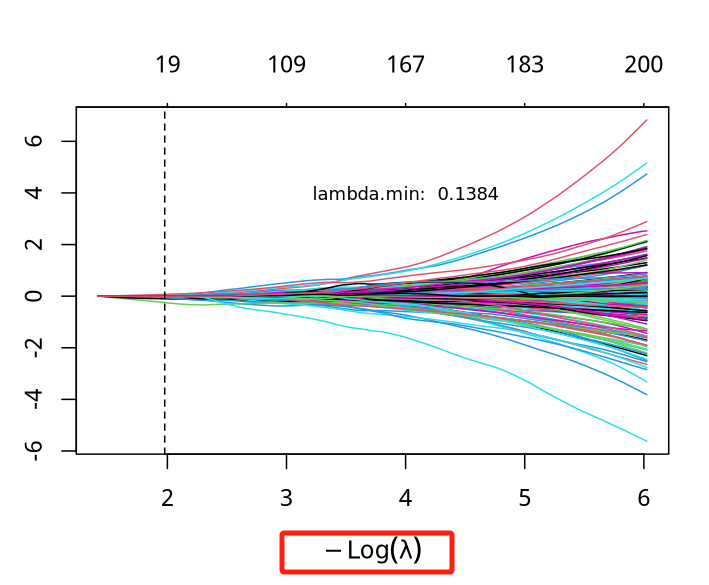
Lasso回归分析交叉验证曲线(均方误差随λ的变化而变化):
png(paste0('./results/Figure3/lasso/S5B-cvfit.png'), width = 5,height = 5,res=1200,units="in")
plot(cvfit)
text(3,210, labels = paste0("lambda.min: ", round(cvfit$lambda.min,4)),col = "red",cex = 0.75)
text(3,200, labels = paste0("lambda.1se: ", round(cvfit$lambda.1se,4)),col = "black",cex = 0.75)
dev.off()
「横坐标:」
上横坐标是对应模型所需变量数目,从右到左逐渐减少
下横坐标轴是- log λ,也称为lambda惩罚系数的对数负数
「纵坐标:」均方误差(MSE, mean squared error),指的是计算模型的预测值与真实值的差异程度,每个MSE都有对应的误差棒,以表示MSE的95%置信区间。Y轴越小说明方程的拟合效果越好。
「虚线:」一般会有两条虚线,右边虚线是λ.min,指示最小MSE对应的横坐标。偏差最小时的λ ,代表在该λ 取值下,模型拟合效果最高。靠左的虚线λ.1se,指距离λ.min一个标准误差的位置,这个包含的变量更少,模型更简洁。在实际应用中若λ.min与 λ.1se的MSE差别不大,可考虑更简洁模型;若差别较大,则根据研究目的选择更准确的 λ.min或更简洁的λ.1se~
Step 3: Best Lasso model
这里选择了cvfit$lambda.min 来构建最佳模型
# Step 3: Best Lasso model
# 选择 cvfit$lambda.min
cvfit$lambda.min
lasso_best <- glmnet(x = x.train,
y = y.train,
alpha = 1,
lambda = cvfit$lambda.min,
nfolds = 10,
family = "cox")
gene.coef <- as.matrix(round(coef(lasso_best), 4))
coef.lasso <- gene.coef[which(gene.coef[, 1] != 0), ]
coef.lasso <- data.frame(genes=names(coef.lasso),
coefficient=as.numeric(coef.lasso)) %>% arrange(-coefficient)
write.xlsx(coef.lasso, "./results/Figure3/lasso/2.coef_PDAC.Pro.xlsx", rowNames = F,colNames = T,overwrite = T)
coef.lasso
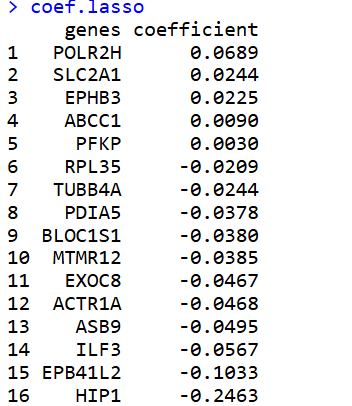
计算模型打分,并根据打分对样本按照中位数score分组:
## Lasso score
y <- as.data.frame(y.train) %>%
mutate(LassoScore=predict(lasso_best,s=cvfit$lambda.min,newx=x.train)) %>%
mutate(LassoScore=as.numeric(LassoScore), ID=rownames(y.train))
y <- y %>% mutate(LASSO.level=ifelse(LassoScore>median(LassoScore),"High" ,"Low")) %>%
mutate(LASSO.level=factor(LASSO.level,levels = c( "Low","High"))) ## cutoff : median
head(y)
write.xlsx(y,paste0('./results/Figure3/lasso/3.Lasso_Score.xlsx'),rowNames = T,colNames = T,overwrite = T)

Step 4: Visualization
对模型中的基因以及对应的系数进行可视化:
# Step 4: Visualization
## bar plot : gene cofficient
df.coef <- coef.lasso
rownames(df.coef) <- df.coef$genes
df.coef <- df.coef %>% transmute(gene=genes,cor=coefficient) %>%
mutate(gene=paste0(gene," (",cor,")")) %>%
arrange(cor) %>%
mutate(gene=factor(gene,levels = gene)) %>%
mutate(group=ifelse(cor>0,"A","B"))
p1 <- ggplot(df.coef, aes(gene, cor, fill = group)) +
geom_bar(stat = 'identity') +
ylab('coefficient') +
xlab('') +
guides(colour=F,fill=F) +
theme_bw(base_size = 12) +
scale_fill_manual(values = brewer.pal(9,'Set1')) +
theme(panel.grid =element_blank(),
axis.text = element_text(colour = 'black'),
axis.text.x = element_text(size = 8)) +
coord_flip()
p1
ggsave("./results/Figure3/5A-Lasso_cofficient_barplot.png",p1,width = 3,height = 4)
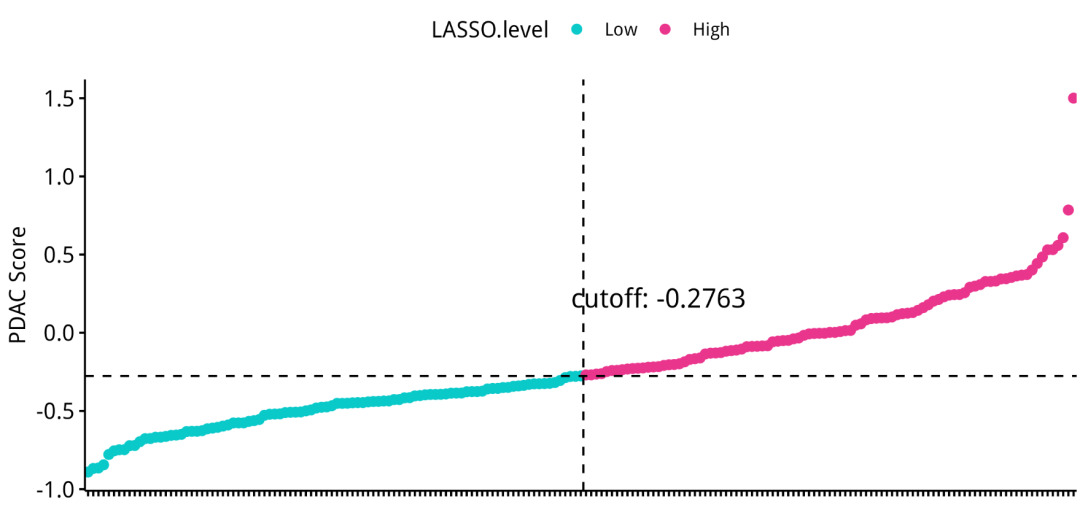
每个样本中的打分排序气泡图:
## dot plot : lasso score
df.score <- y
df.score <- df.score %>%
mutate(samples=rownames(df.score)) %>%
arrange(LassoScore) %>%
mutate(samples=factor(samples,levels = samples))
head(df.score)
p2 <- ggscatter(df.score, x="samples", y="LassoScore", color = "LASSO.level",
palette = c("#0ac9c9","#ea358d")) +
geom_hline(yintercept=median(df.score$LassoScore), linetype=2,color='black') +
geom_vline(xintercept=median(1:nrow(df.score))+0.5, linetype=2,color='black') +
annotate('text',
x=median(1:nrow(df.score))+15,
y=median(df.score$LassoScore)+0.5,
label=paste0('cutoff: ', round(median(df.score$LassoScore),4)),size=5, color='black') +
ylab('PDAC Score') +
xlab('') +
theme(axis.text.x = element_blank())
p2
ggsave("./results/Figure3/5A-Lasso_score_dotplot.png",p2,width = 8,height = 4)
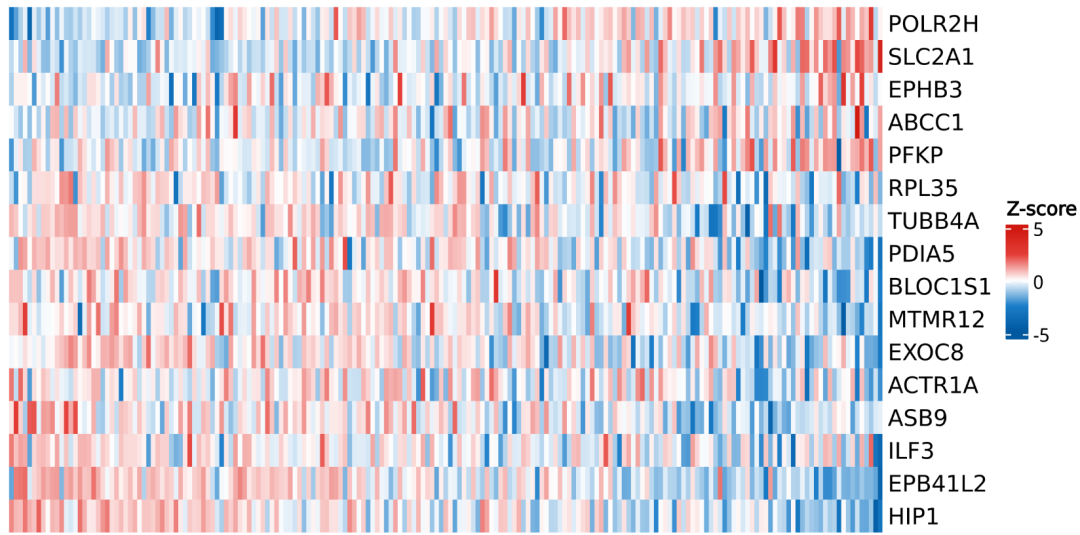
模型中的基因对应的表达矩阵热图:
## Heatmap : protein abundance
heat.data <- t(x.train)
heat.data <- heat.data[rev(rownames(df.coef)),row.names(df.score)]
heat.data <- t(scale(t(heat.data)))
head(heat.data)
png("./results/Figure3/5A-Lasso_gene_heatmap.png",width = 8,height = 4,res=1200,units="in")
p3 <- Heatmap(heat.data, name = "Z-score",
col = colorRampPalette(c("#00599F","#1c7ec9","#FFFFFF",
"#e53f39","#D01910"))(100),
cluster_rows = F,cluster_columns = F,show_column_names = F)
draw(p3)
dev.off()
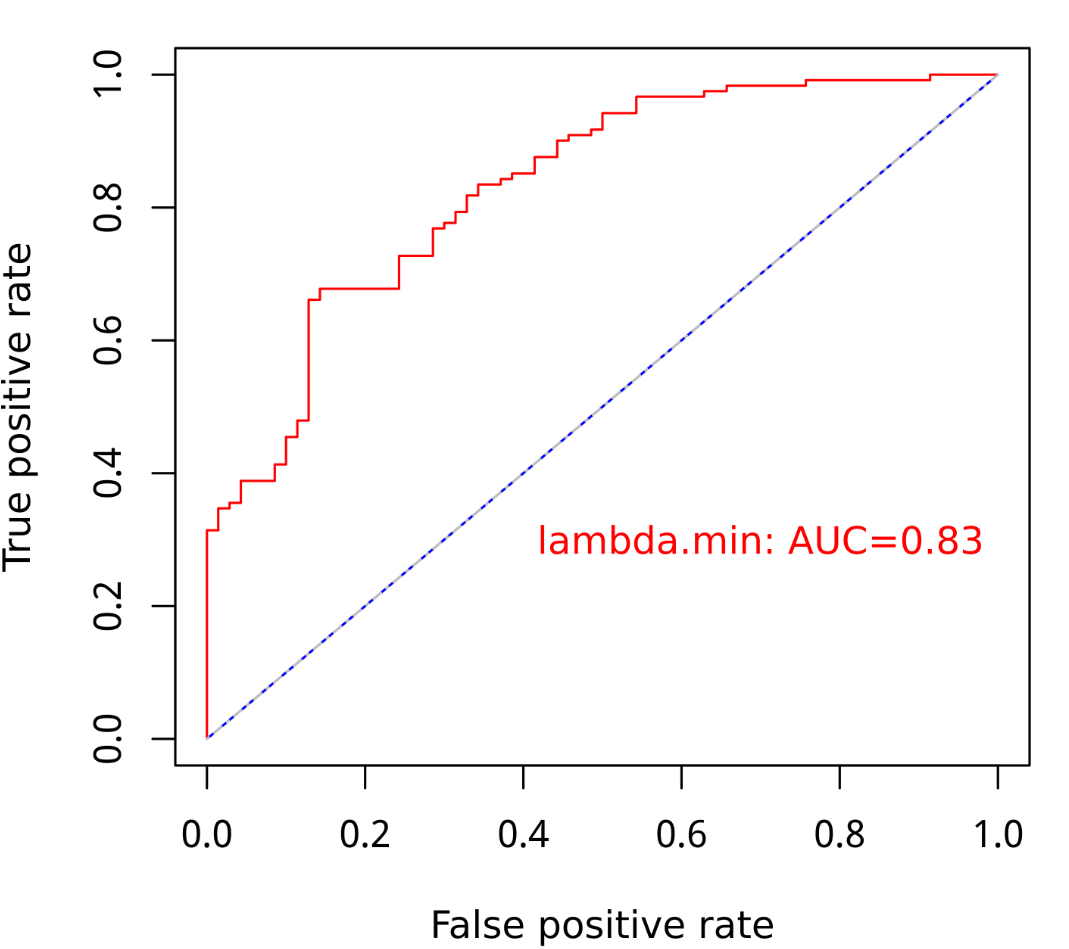
Step 5: ROC曲线绘制
ROC曲线:看下模型的预测能力
# Step 5: receiver operating characteristic (ROC)
# performance of model
lasso.prob <- predict(cvfit, newx=x.train , family = "cox",
s=c(cvfit$lambda.min, cvfit$lambda.1se) )
re <- cbind(as.data.frame(y.train),lasso.prob) %>% as.data.frame()
colnames(re) <- c('time','status','prob_min','prob_1se')
# model: lambda.min
pred_min <- prediction(predictions=re$prob_min, labels=re$status) # predictions: containing the prediction. labels: true class labels
perf_min <- performance(pred_min,"tpr","fpr")
auc_min <- performance(pred_min,"auc")@y.values[[1]]
# mocel: lambda.1se
pred_1se <- prediction(re$prob_1se, re$status)
perf_1se <- performance(pred_1se,"tpr","fpr")
auc_1se <- performance(pred_1se,"auc")@y.values[[1]]
## plot
png(paste0('./results/Figure3/lasso/S5C-ROC.png'), width = 5,height = 5,res=1200,units="in")
plot(perf_min,colorize=F, col="red")
plot(perf_1se,colorize=F, col="blue",add = T)
lines(c(0,1),c(0,1), col = "gray", lty = 4 )
text(0.7,0.3, labels = paste0("lambda.min: AUC=", round(auc_min,2)),col = "red")
dev.off()
自己的数据非独立验证,ROC结果0.83还行吧。
Step 6: Multivariate analysis
看一下打分是不是独立预后因子:
# Step 6: Multivariate analysis
# data
y_forest <- y %>% left_join(cli.pdac , by =c('ID'='ID'))
y_forest <- y_forest %>% dplyr::select( c('time','status','LASSO.level',
'age','gender','CA125',
'CA19_9','CEA','Smoking',"Drinking",'AJCC_V8',
'Censoring_chemo'))
y_forest <- y_forest %>% dplyr::rename(LassoLevel=LASSO.level,
Age=age,Gender=gender,
CA199=CA19_9,
Adjuvant_therapy=Censoring_chemo,
AJCC_stage=AJCC_V8)
y_forest <- y_forest %>% mutate(Gender=factor(Gender,levels = c("Female","Male")))
y_forest[1:6,1:10]
# AJCC_stage
table(y_forest$AJCC_stage)
a <- y_forest$AJCC_stage
b <- recode(a, Ia = "I", Ib = "I",IIa= "II", IIb= "II",III="III",IV="IV")
y_forest$AJCC_stage <- factor(b,levels = c("I","II","III"))
# coxph
cfit <- coxph(Surv(time,status)~LassoLevel+Age+Gender+CA125+CA199+
CEA+Smoking+Drinking+AJCC_stage+Adjuvant_therapy,
data = y_forest)
# Forest plots
p <- forest_model(cfit,
format_options = forest_model_format_options(colour = brewer.pal(9,'Set1')[2], shape = 16,text_size = 15,point_size = 2.5, banded = T),
factor_separate_line = F )
ggsave(paste0("./results/Figure3/S5D-multivariate_forest.png"), p, width = 8, height = 6)
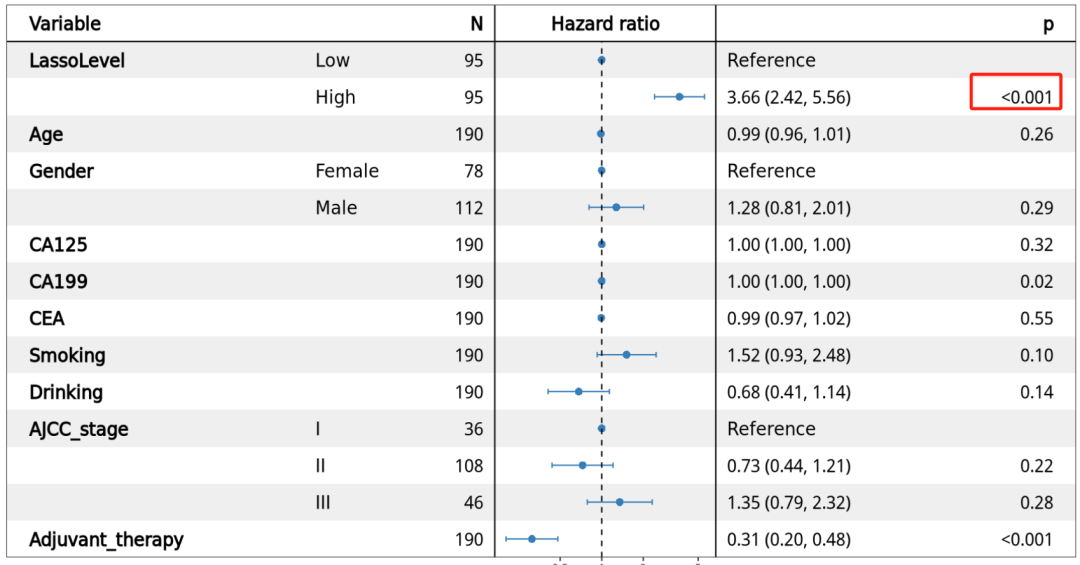
而且打分高与不好的预后相关。
今天分享到这~
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-10-09,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号