物理计算 vs AI 预测:谁才是蛋白质-配体结合亲和力预测的真正胜者?

物理计算 vs AI 预测:谁才是蛋白质-配体结合亲和力预测的真正胜者?

DrugIntel
发布于 2026-05-14 18:00:48
发布于 2026-05-14 18:00:48

文献速递 | ByteDance AI Drug Discovery × Anew Therapeutics 原文标题:Physics-Based vs AI-Based Free Energy Prediction for Protein-Ligand Potency: Public Benchmarks and Internal Project Evidence 发表平台:ChemRxiv(预印本,2026年4月28日) DOI:10.26434/chemrxiv.15002526/v1
一句话摘要
在公开基准上,AI 方法表现亮眼,但在真实药物发现的全前瞻性场景(de novo 分子、分布外化学结构)中,物理自由能计算仍不可替代——AI 的排序能力几近崩溃,而 AnewFEP 依然保持有效区分度。
1. 研究背景与核心问题
1.1 为什么结合亲和力预测如此重要?
蛋白质-配体结合自由能(ΔG)是药物分子"有效性"的热力学核心指标。在先导化合物优化阶段,每一轮设计-合成-测试(DMTA 循环)耗资巨大,而计算方法的作用正是在实验之前对候选分子进行预筛,缩短优化周期、降低试错成本。
预测精度通常以 kcal/mol 为单位衡量。业界经验法则认为:
- • RMSE < 1.0 kcal/mol:接近实验噪声,具备强决策参考价值
- • RMSE 1.0–1.5 kcal/mol:良好,可用于排序和优先级筛选
- • RMSE 1.5–2.0 kcal/mol:中等,仅供参考
- • RMSE > 2.0 kcal/mol:可靠性存疑,需谨慎使用
即使 0.5 kcal/mol 的精度差异,在大规模扰动网络中也会对决策产生显著影响。
1.2 两大技术路线的本质区别
物理方法(RBFE) 通过统计力学严格计算自由能差:
采用分子动力学(MD)模拟 + 炼金变换(alchemical transformation)在一系列中间 λ 窗口之间逐步"变形"配体 A 为配体 B,利用 BAR/MBAR 估算自由能差。核心优势在于原理正确性:只要力场精确、采样充分,结果在热力学上是严格的。
AI 方法 则从数据中直接学习结构-活性关系。按输入类型简单分为两类:
类别 | 代表模型 | 输入 | 核心机制 |
|---|---|---|---|
结构打分函数 | PBCNet2、PIGNet2 | 预定义的 3D 蛋白-配体复合物位姿 | 学习原子间相互作用的空间特征表示 |
结构预测基础模型 | Boltz-2 | 蛋白序列 + 配体 SMILES | 端到端预测结构,同时输出亲和力置信分数 |
1.3 本文要回答的两个核心问题
- 1. 精度决定因素:现代 RBFE 工作流的精度与鲁棒性主要由哪些因素决定?为什么 FEP+ 的性能难以被其他实现所复现?
- 2. AI 的真实边界:在什么条件下,AI 方法可以补充乃至替代物理 RBFE 计算?特别是面对分布外(OOD)的全新化学结构时?
2. AnewFEP:一套工程驱动的 RBFE 新工作流
2.1 技术架构概览
AnewFEP 是本文的核心方法贡献,基于开源 GROMACS 引擎构建,主要由以下模块组成:
输入准备
├── 蛋白质结构处理(Amber ff14SB 力场)
├── 配体参数化(AnewFF 力场)
├── 原子映射(atom mapping)
└── 扰动图构建(perturbation graph generation)
↓
模拟执行
├── 系统构建 + λ 窗口准备
├── 约束能量最小化(50,000 步)
├── NVT 弛豫(400 ps,1 fs 步长)
├── NPT 平衡(100 ps → 600 ps,2/4 fs 步长)
└── REST2 生产模拟(20 ns × 3 独立运行)
↓
分析模块
├── BAR/MBAR 自由能估算(cinnabar)
├── 相空间重叠质量评估
├── 自由能收敛性检验
├── 蛋白-配体相互作用分析(ProLIF)
└── 配体扭转角分布分析(MDAnalysis)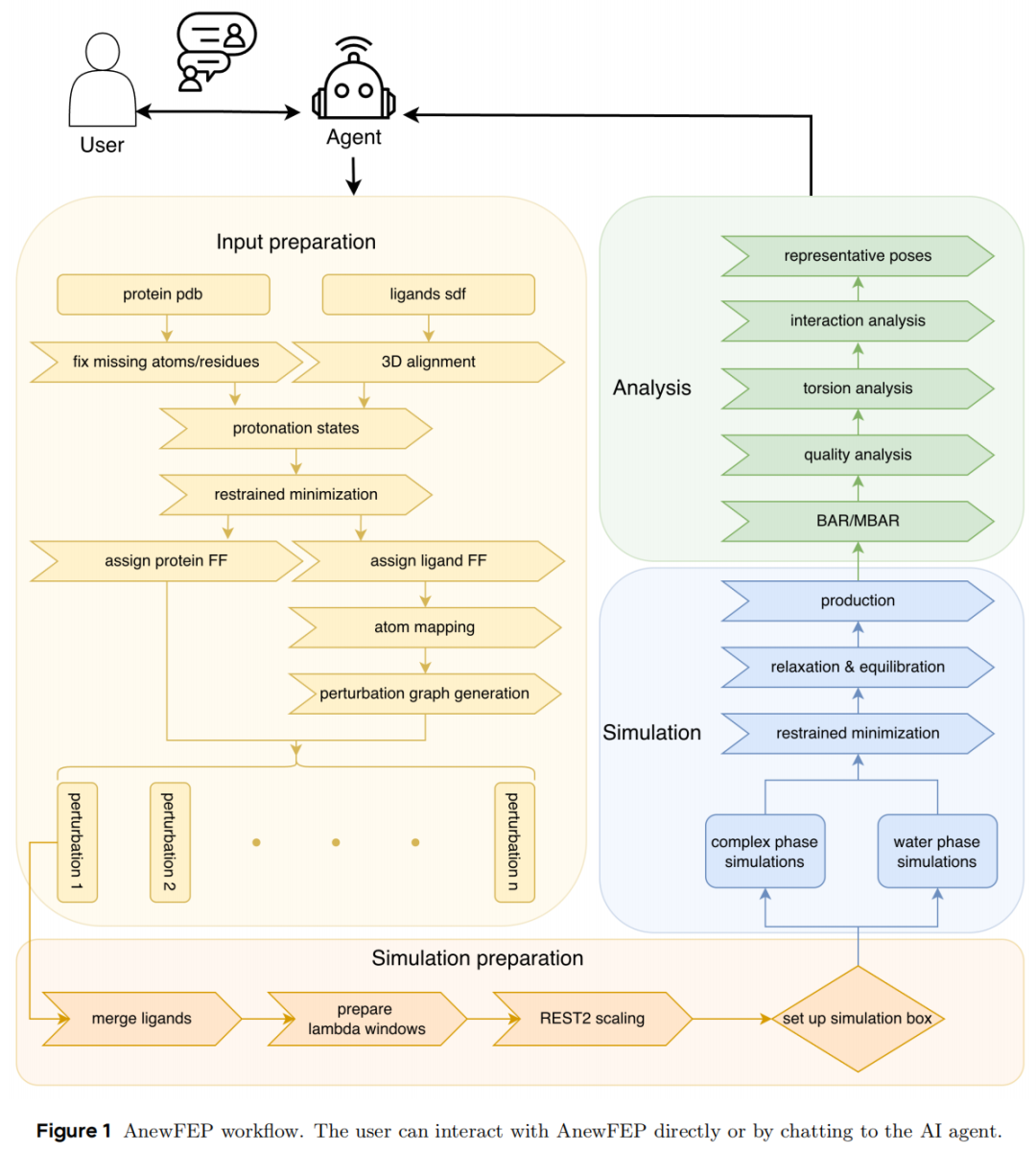
2.2 AnewFF 力场
AnewFF 基于字节跳动此前发布的 ByteFF 机器学习力场,通过对内部多个药物发现项目的系统误差分析,进一步精细化了非键相互作用参数(特别是 van der Waals σ 和 ε 参数)。
相比传统半经验力场(如 GAFF2、OPLS),ML 衍生力场在化学空间覆盖度和参数一致性上有天然优势,但仍需针对特定官能团(如磺酰基、杂环等)进行局部校准——这正是本文案例研究的核心主题之一。
2.3 工程层面的关键改进
本文在 GROMACS-based FEP 实现中发现并修复了多个长期存在的问题:
(1) GPU 混合精度数值不稳定性
在混合精度 FEP 模拟中,GPU 求和操作的非确定性归约顺序导致 A→A 自由能差非零(应严格为 0)。解决方案:引入双精度 GPU 归约方案,消除第七位有效数字的误差积累。
(2) 核心骨架跃迁的软键(soft-bond)处理
对于 core-hopping 类型的炼金变换,标准简谐势在 λ→0 时趋向奇点,导致模拟崩溃。团队开发了一种新型软键势:
在确保 λ=0 时数值稳定的同时,与商业实现的专利方案相区别,并支持大环分子的稳定采样。
(3) 独立 λ 缩放控制
扩展 GROMACS 的炼金变换,允许对不同原子组(配体 A 原子、配体 B 原子、公共核心原子)独立控制 λ 缩放,显著提升相空间重叠度和炼金变换效率。
(4) 多时间步长(MTS)GPU 优化
针对 AnewFEP 的模拟体系优化了 MTS 的 GPU 实现,将短程非键相互作用(快变量)与长程静电(慢变量)分离计算,在不损失精度的前提下显著加速模拟。
(5) Ewald 排除项修正
修正了 GROMACS 长期存在的一个 Ewald 排除处理 bug:在邻居列表截断之外的扰动排除原子对未正确计算长程静电修正,这对 core-hopping 变换的准确性至关重要。
(6) 氢质量重分布(HMR)
精细化 HMR 的实现与 AnewFEP 工作流的集成,将积分步长提升至 4 fs,生产效率提升约 2 倍。
2.4 AI Agent 集成
AnewFEP 提供对话式 AI agent 接口,支持用户通过自然语言指令完成任务提交、数据分析和分子过滤(例如:"筛选与 His48 形成氢键且预测 ΔG < -9 kcal/mol 的分子"),降低使用门槛并加速迭代。
3. 评测体系与基准数据集
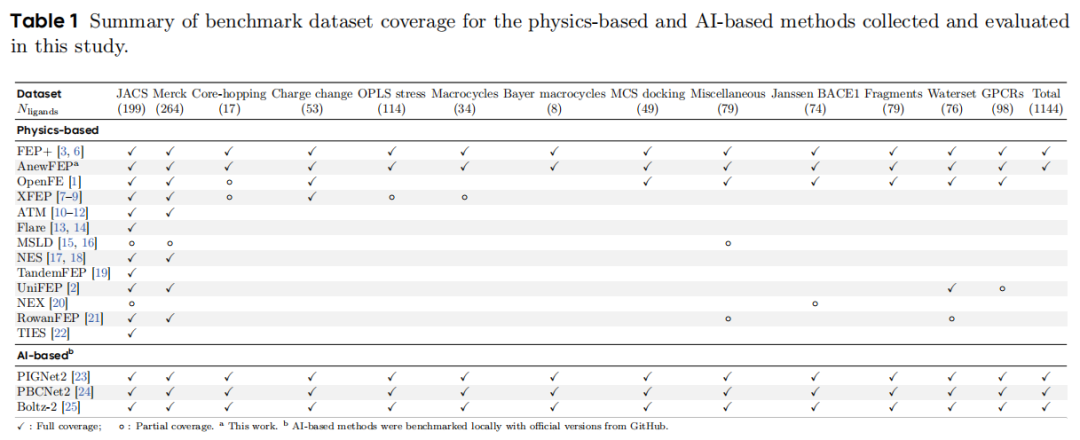
3.1 数据集构成
本文采用的公开基准来自 Ross et al. (2023) 的工作,涵盖 1144 个配体,分属 14 个子集:
子集 | 配体数 | 主要挑战 |
|---|---|---|
JACS | 199 | 经典 R 基团替换,较基础 |
Merck | 264 | 多靶点综合,行业标杆 |
Core-hopping | 17 | 骨架跳跃,拓扑变化大 |
Charge change | 53 | 净电荷改变,静电处理难 |
OPLS stress | 114 | 专门测试力场非键参数 |
Macrocycles | 34 | 大环构象采样困难 |
Bayer macrocycles | 8 | 大环体系 |
MCS docking | 49 | 依赖对接位姿质量 |
Miscellaneous | 79 | 混合体系 |
Janssen BACE1 | 74 | β-分泌酶抑制剂 |
Fragments | 79 | 片段类配体 |
Waterset | 76 | 需要显式水分子处理 |
GPCRs | 98 | G 蛋白偶联受体,膜蛋白 |
3.2 评价指标
- • 主要指标:基于扰动图的成对 ΔΔG RMSE(pairwise RMSE),消除不同图拓扑对结果比较的影响
- • 辅助指标:绝对亲和力 ΔG 的 R²
- • 加权平均:按各系统化合物数量加权,与 Ross et al. 方法一致
4. 主要结果:基准测试全景
4.1 物理方法整体排名
在覆盖最广的综合基准上,FEP+ 和 AnewFEP 以显著优势领先:
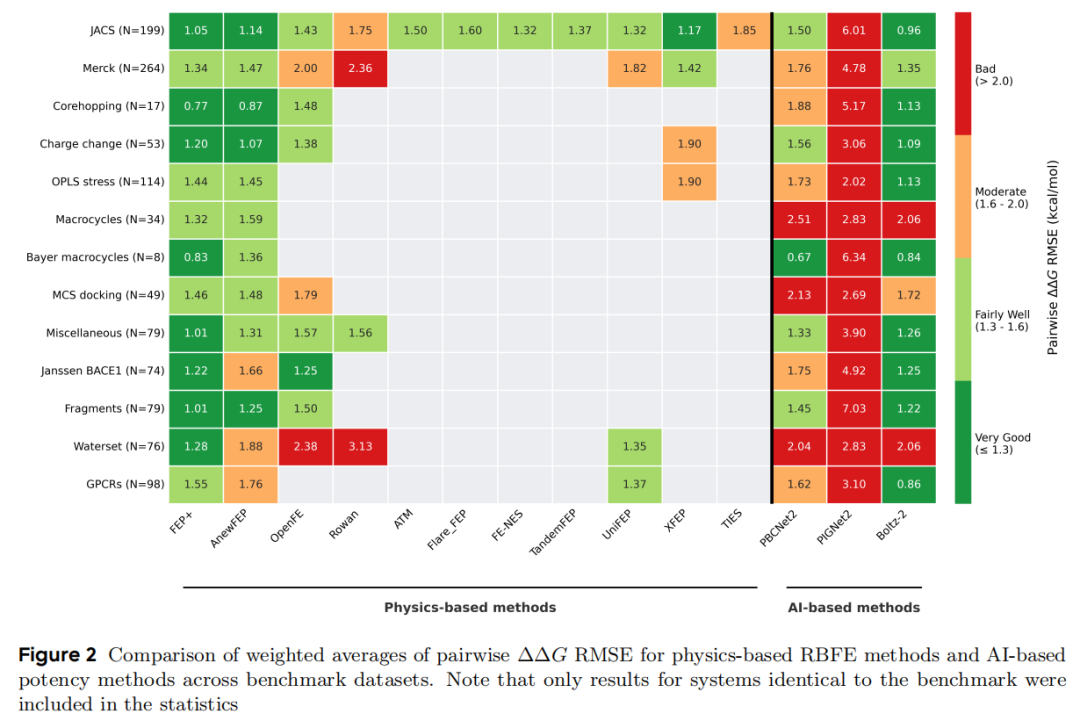
关键观察:
- • JACS 数据集因扰动局部性强,大多数方法均可达到 1.5 kcal/mol 以下,区分度较低
- • Merck、OPLS stress 和 Core-hopping 是真正的"试金石",多种方法在此 RMSE 超过 2.0
- • AnewFEP 在 Waterset 上误差偏高(1.88 vs FEP+ 的 1.28),原因是缺少大正则蒙特卡洛(GCMC)水分子采样模块
4.2 AI 方法在公开基准上的表现
方法 | 整体 RMSE (kcal/mol) | JACS | Merck | GPCRs | 核心局限 |
|---|---|---|---|---|---|
Boltz-2 | 1.25 | 0.96 | 1.35 | 0.86 | 依赖位姿质量;OOD 泛化差 |
PBCNet2 | 1.50 | 1.50 | 1.76 | 1.62 | 输入位姿误差直接传播 |
PIGNet2 | 3.39 | 6.01 | 4.78 | 3.10 | 整体表现不稳定 |
Boltz-2 的公开基准 RMSE 与 FEP+ 持平,乍看令人惊艳。然而后续前瞻性实验将揭示这一数字的局限性。
4.3 FEP+ 长期领先的三个工程假说
作者系统归纳了 FEP+ 超越其他开源实现的三个核心原因:
① 力场的迭代积累
OPLS 力场经历数十年、数代更新,大量针对类药分子空间(drug-like chemical space)的参数经验被编码进去,提供了对多数官能团的可靠基线描述。
② Dummy 原子处理的严格分解性
炼金变换中,dummy 原子(消失/出现的原子)对配分函数的贡献需要满足"可分解"条件,才能在自由能差中相消:
中的贡献相消
若内坐标定义、力场参数和原子映射不一致,dummy 原子贡献将引入系统性偏差,且极易被误认为是力场误差或采样不足。FEP+ 在这一细节上经过长期打磨。
③ 针对特殊场景的专属协议
FEP+ 为净电荷变化、core-hopping、共价配体、片段连接、显式水分子置换等场景提供了专属工作流,而通用实现往往在这些边界条件上失效。
5. 误差来源解剖:四个深度案例研究
5.1 案例一:磺酰基氧的 van der Waals 参数——HIF-2α 体系
系统背景:HIF-2α(缺氧诱导因子-2α)是一个重要的肿瘤靶点,其配体系列对 RBFE 计算构成了显著挑战。多数方法在此数据集上表现不佳(XFEP 1.9、UniFEP 2.2、OpenFE 2.3、Rowan 2.7 kcal/mol)。
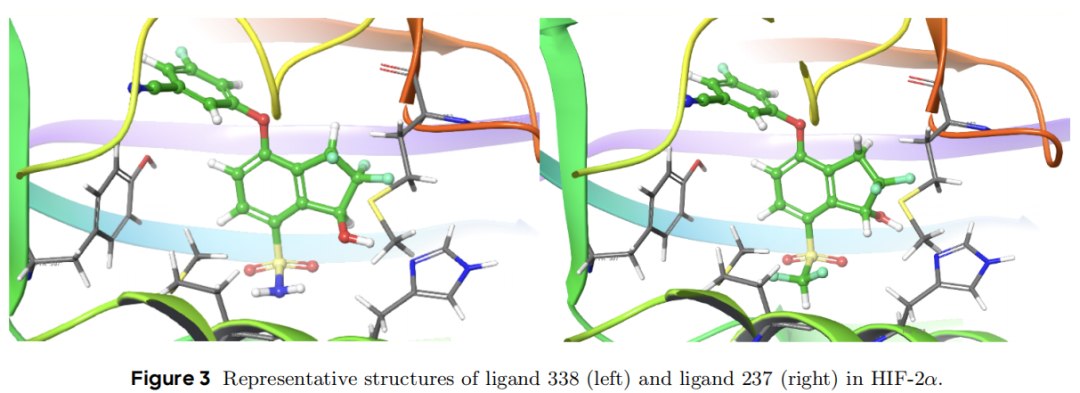
误差溯源:以 338→237 扰动为例分析:
参数 | 实验 ΔΔG | 计算 ΔΔG | 误差 |
|---|---|---|---|
原始 σ = 3.04 Å | +1.20 | −3.00 | 4.20 kcal/mol |
σ = 3.10 Å | +1.20 | −2.56 | 3.76 kcal/mol |
σ = 3.30 Å | +1.20 | −1.86 | 3.06 kcal/mol |
σ = 3.50 Å | +1.20 | −1.16 | 2.36 kcal/mol |

物理机制:原始 σ 值低估了磺酰基氧的有效排斥体积,导致配体 237 在蛋白质结合位点形成不真实的近距离接触,虚假过稳定化。关键证明:单独调整 ε(势阱深度)无法修复误差,说明根本原因是位阻排斥而非色散强度。
改进效果:将 SO₂ 氧的 σ 从 3.04 Å 精细化至约 3.30–3.50 Å 后,HIF-2α 体系整体 pairwise RMSE 从 2.1 降至 1.5 kcal/mol,改善幅度传导至整个扰动网络。
方法论启示:针对特定官能团的局部非键参数校准,可以在不重新训练整体力场的前提下,实现系统级精度提升。这也说明了为什么 ML 衍生力场仍需要领域专家进行靶向优化。
5.2 案例二:扭转势垒的蝴蝶效应——BACE1 cr2 压力测试
系统背景:OPLS 压力测试集(stress test)专为暴露力场非键和成键参数缺陷而设计,是区分不同 RBFE 实现真实能力的重要标尺。BACE1 cr2 子集中的 11→13 扰动是典型失效案例。
失效分析:
- • 实验值:ΔΔG_exp ≈ 0 kcal/mol(近似等能变换)
- • 原始力场预测:ΔΔG ≈ −2.0 kcal/mol(人为过稳定化)
- • 化学差异:配体 11 与 13 仅相差**一个原子替换(C→N)**于芳环
对 2–1–42–14 二面角的扭转势能曲线比较显示:原始力场在该二面角上存在夸大的旋转势垒,导致两个配体在溶剂和蛋白质相中的构象集合均发生系统性偏移。
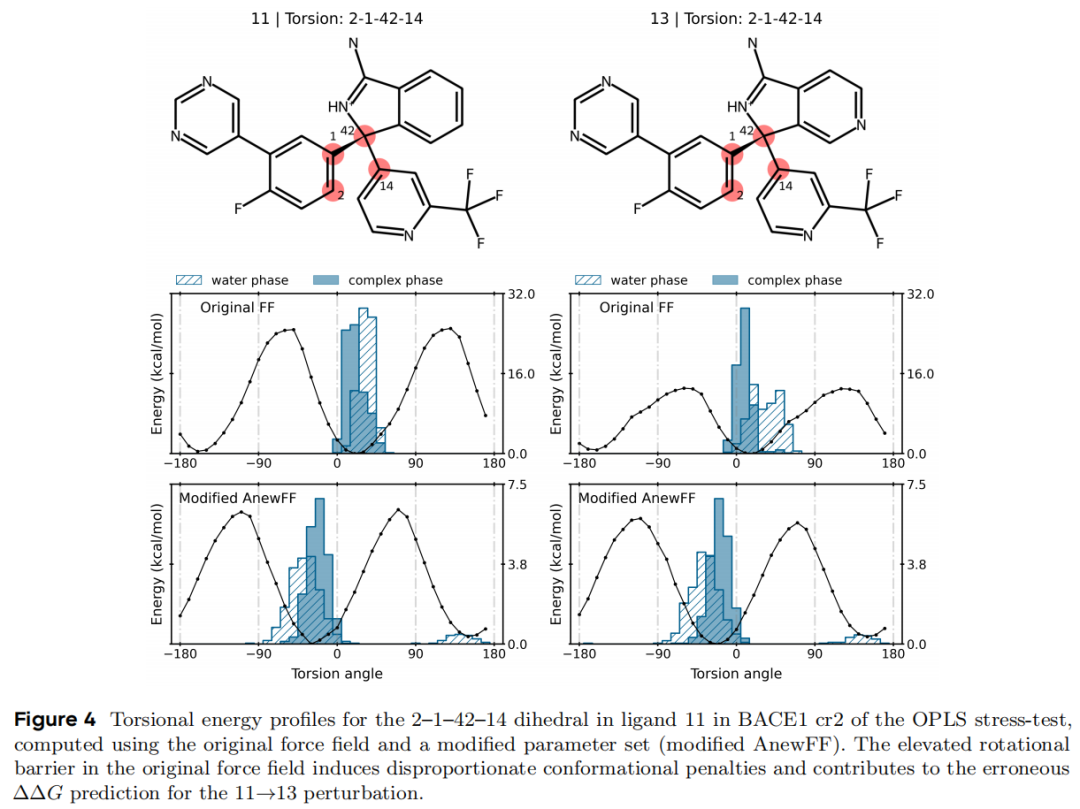
深层含义:在实验信号弱(ΔΔG ≈ 0)的扰动边上,哪怕较小的力场偏差也会被放大为多 kcal/mol 的假阳性误差。这类误差往往比大误差更危险,因为它会在本应被排除的分子上浪费合成资源。
修复策略:更新 AnewFF 中对应二面角的傅里叶展开参数,使得两个配体的扭转势能曲线在物理上合理(具有相似的势阱深度和势垒高度),从而将计算误差从 2.0 降至 0.5 kcal/mol 以下。
5.3 案例三:蛋白质结合位点漂移——FXa Set 6
系统背景:FXa(凝血因子 Xa)是重要的抗凝靶点,Set 6 属于 OPLS 压力测试集中的高难度子集,原始力场给出 pairwise RMSE = 2.41 kcal/mol。
关键扰动:21084 → 20524_2j95_cys
方案 | 计算 ΔΔG | 实验 ΔΔG | 绝对误差 |
|---|---|---|---|
无约束(原始) | −0.763 | −3.95 | 3.19 kcal/mol |
加入主链二面角约束 | −2.66 | −3.95 | 1.29 kcal/mol |
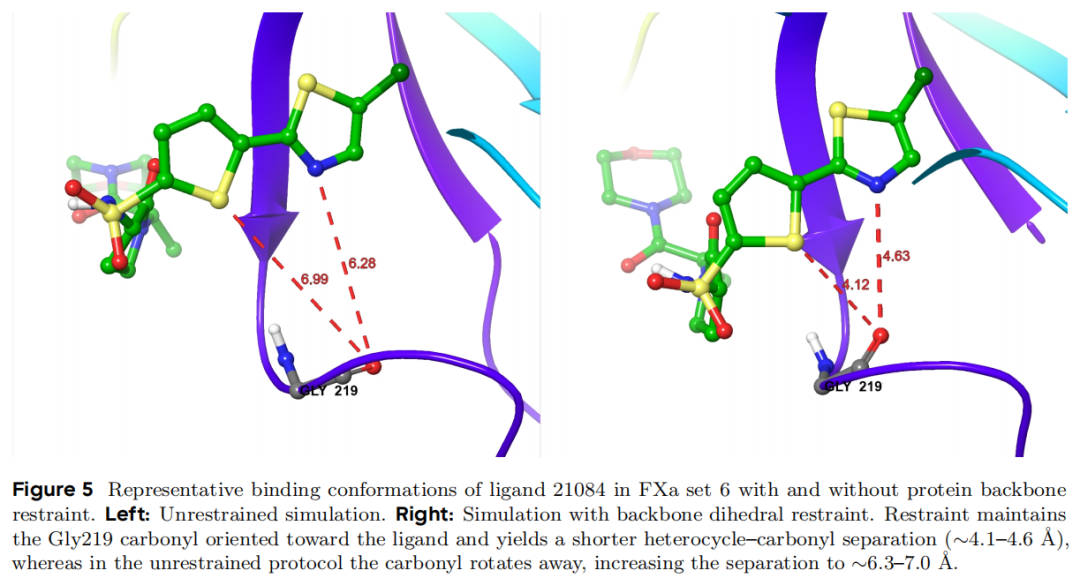
结构机制:Gly219 的主链羰基(C=O)是该结合位点中与配体发生关键相互作用的残基。在无约束模拟中,该羰基在 20 ns 尺度上自发旋转偏离,导致异环-羰基间距从约 4.1–4.6 Å 增大至 6.3–7.0 Å,相互作用基本消失。
施加主链二面角约束后,Gly219 羰基维持朝向配体的构象,正确重现了结合位点几何形状。整体 RMSE 从 2.41 降至 1.57 kcal/mol,改善约 0.84 kcal/mol。
普适性意义:这个案例揭示了 RBFE 中一个被系统性低估的误差来源——蛋白质结合位点的微构象漂移。20 ns 的标准生产时间对于蛋白质主链的某些慢速运动而言仍嫌不足,而这些运动对亲和力预测有直接影响。约束策略需要针对每个靶点的结构特征仔细设计,避免过度约束影响采样有效性。
5.4 案例四:λ 调度设计与构象陷阱——JNK1 芳环翻转
系统背景:JNK1(c-Jun N 端激酶 1)的配体结合口袋空间紧凑,某些配体存在甲氧基取代苯环的旋转异构体(rotamer),即芳环翻转问题。
问题量化:17124-1 → 17124-1_flip(两者仅甲氧基朝向不同,理论 ΔΔG = 0)
λ 调度 | 计算 ΔΔG | 理论值 | 误差 |
|---|---|---|---|
原始调度 | 4.10 kcal/mol | 0 | 4.10 kcal/mol |
交互-分离调度 | 0.48 kcal/mol | 0 | 0.48 kcal/mol |
跨所有 17 对翻转扰动的统计结果:
指标 | 原始调度 | 交互-分离调度 |
|---|---|---|
平均 |ΔΔG|(kcal/mol) | 2.27 | 0.49 |
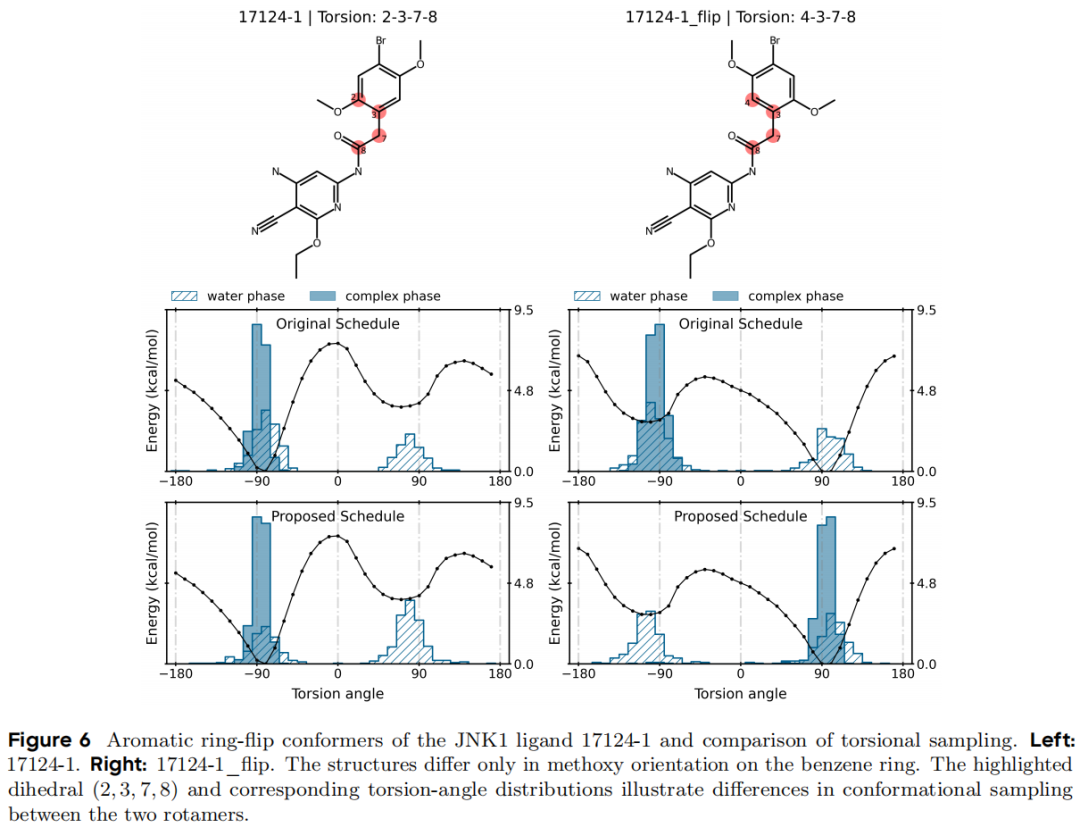
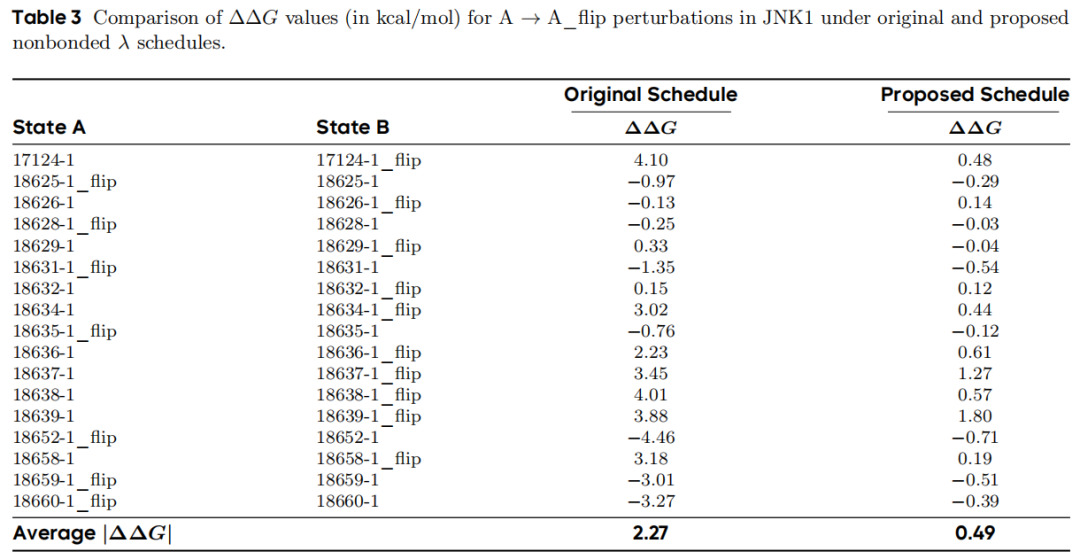
机制解析:在原始调度中,A 和 B 状态的非键参数在中间 λ 窗口中同时存在,导致配体与蛋白质之间持续的非键耦合,形成动力学陷阱——配体被"锁定"在某一构象。
交互-分离 λ 调度的改进思路:在中间 λ 区间,先完全去除 A 状态的非键相互作用,再逐渐建立 B 状态的,从而在"真空"状态中允许配体自由重组。数学上通过自适应软核参数 α_LJ 实现:
与 FEP+ 通过在扰动网络中显式加入多个旋转构象节点的方案相比,λ 调度方案无需预先识别动力学陷阱,通用性更强。
6. 前瞻性内部研究:AI 的真实边界
6.1 实验设计
这是本文最有价值的贡献之一。研究团队在内部真实药物发现项目的 de novo 分子上同时运行 AnewFEP 和 Boltz-2,这些分子:
- • 完全不在任何公开数据集中
- • 由内部团队从头设计,代表真实的先导化合物优化场景
- • 存在训练分布外(OOD)的化学结构和结合模式
6.2 结果对比
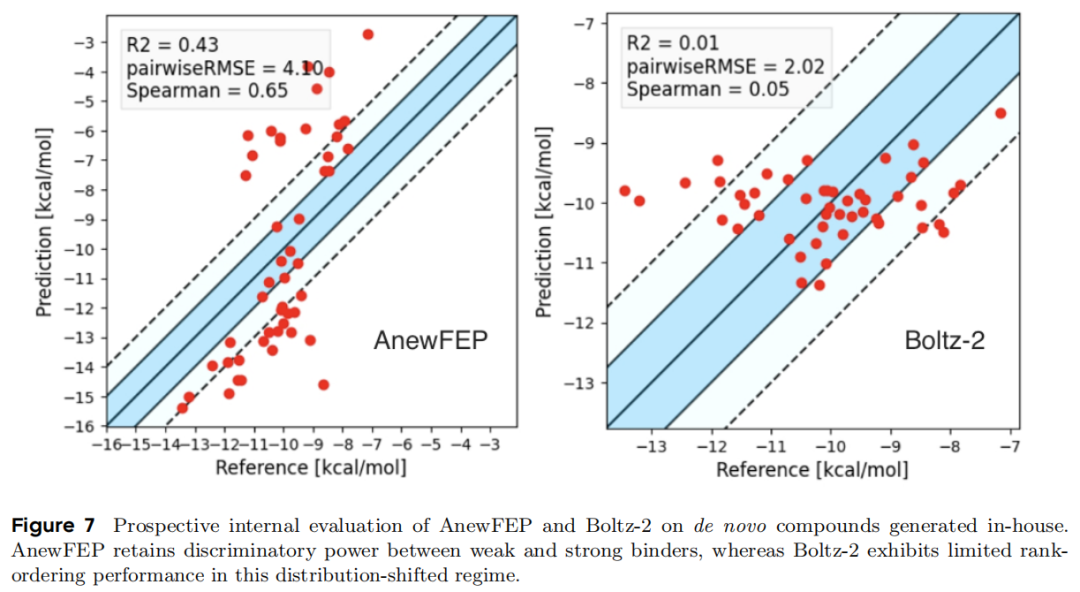
指标 | AnewFEP | Boltz-2 |
|---|---|---|
R² (ΔG 相关性) | 0.43 | 0.01 |
pairwise RMSE | 4.10 kcal/mol | 2.02 kcal/mol |
Spearman 相关系数 | 0.65 | 0.05 |
排序能力 | ✅ 有效区分强弱结合物 | ❌ 几近随机 |
注意:Boltz-2 的 pairwise RMSE 看似低于 AnewFEP,但 R² 和 Spearman 相关系数揭示其预测结果与实验值几乎无相关性,较低 RMSE 源于预测值聚集在均值附近(无区分度),而非真正的准确预测。
6.3 失败模式分析
Boltz-2 的失败根源:
- 1. 位姿敏感性:Boltz-2 的亲和力预测以其生成的蛋白-配体结构为条件(conditional generation)。对于训练集之外的新化学骨架和结合模式,结构预测本身的误差直接传播至亲和力估算,形成误差级联。
- 2. 模式识别 vs 热力学推理:一个揭示性实验——在 PDB 3R58 体系中,磷酸辅因子对准确的 FEP 计算至关重要(显著影响结合位点的静电环境),但 Boltz-2 无论有无辅因子,预测结果几乎相同。这表明模型的预测更多依赖训练数据中的统计模式,而非对具体热力学环境的物理感知。
- 3. 分布外泛化困难:这与近期多项研究的发现一致——蛋白质-配体共折叠模型在 OOD 场景中普遍表现下降,其"记忆力"强于"推理力"。
AnewFEP 的鲁棒性来源:物理 RBFE 从第一性原理出发,估算的是在用户指定结合构型周围的 MD 采样所代表的热力学系综。只要初始结合位姿合理(可由对接或晶体结构提供),自由能差就能在力场精度范围内可靠估算,与分子是否"见过"无关。
7. 综合讨论:物理方法领先的深层原因
7.1 精度的三角形:力场 × 采样 × 协议
本文的核心方法论贡献是将 RBFE 精度分解为三个相互依赖的要素:
精度
/ \
力场 采样
\ /
协议- • 力场决定能量函数的正确性(势能面的形状)
- • 采样决定是否充分探索了构象空间(相空间的覆盖度)
- • 协议决定炼金路径的设计合理性(λ 调度、dummy 原子处理、约束策略)
三者缺一不可:完美的力场配合糟糕的采样或协议,同样会导致错误结果。这解释了为什么 FEP+ 的整体优势难以被简单地"换一个更好的力场"所复现。
7.2 AI 方法的合理定位
基于本文及领域内相关工作,AI 方法的实用价值可归纳为:
应用场景 | AI 方法适用性 | 推荐策略 |
|---|---|---|
超大规模虚拟筛选(>10⁶ 分子) | ✅ 高 | 作为第一轮快速过滤 |
骨架类似物的 SAR 分析 | ✅ 中-高 | 可用于初步排序 |
先导化合物精细优化(~10–100 分子) | ⚠️ 低 | 需物理方法验证 |
全新骨架/分布外分子评估 | ❌ 不可靠 | 必须用物理方法 |
新靶点(少量结构数据) | ❌ 极不可靠 | 必须用物理方法 |
7.3 未来方向
论文结尾指出,下一步的进展需要在三个层面协同优化:
- 1. 力场层:小分子与蛋白质力场的联合优化,特别是蛋白质-配体界面的非键参数
- 2. 实现层:数值鲁棒、可重现的开源模拟引擎,适配现代 GPU 架构
- 3. 协议层:能在位姿变异、旋转异构体模糊性和蛋白质柔性等真实不确定性来源下保持稳定的采样和炼金路径设计
前瞻性、多靶点的评测框架将是验证 AI 方法何时可靠替代物理计算的关键基础设施。
关键结论速查
① 物理方法仍是先导优化阶段的核心:在真实前瞻性场景中,AnewFEP 的 Spearman 相关系数 0.65 vs Boltz-2 的 0.05,差距不是一个数量级,而是有无意义之别。
② AnewFEP 证明了开源实现可达 FEP+ 量级:整体 RMSE 1.44 vs 1.25 kcal/mol,差距已接近实验误差范围。
③ RBFE 误差的主导因素按优先级排序:局部力场参数(特别是 vdW σ) > 蛋白质构象控制 > λ 调度设计 > 采样时长。
④ AI 方法的"公开基准幻觉":Boltz-2 在公开基准上的 RMSE 与 FEP+ 持平,但这一数字在真实项目中完全失效,R² 仅为 0.01。
⑤ 实用建议:AI 方法适合大规模初筛;一旦进入关键决策阶段(化合物合成排期、剂量假说制定),物理自由能计算不可省略。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-11,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号