Nat Rev Drug Discov|深度学习与QSAR的融合


2023年12月8日,来自北卡罗来纳大学教堂山分校的Alexander Tropsha、卡内基梅隆大学的Olexandr Isayev等研究人员在Nature Reviews Drug Discovery发表综述文章Integrating QSAR modelling and deep learning in drug discovery: the emergence of deep QSAR。

该文章介绍了人工智能如何赋能定量构效关系(QSAR)研究,并通过详细案例介绍了深度QSAR(deep QSAR)的应用场景,最后对deep QSAR技术的未来进行了展望。
前言
定量构效关系(QSAR)
定量构效关系(Quantitative structure–activity relationship,QSAR)的研究始于1962年,最初用于分析分子性质(如偶极矩、疏水性)与生物活性之间的关系。随着分子表征算法的发展、统计学和机器学习算法的不断优化,以及分子数据集的大小和多样性的增加,QSAR已扩展到不仅预测化合物的生物活性,还包括预测化合物的理化性质(如水溶性、熔点和pKa值)和ADMET特性,并广泛应用于化学和材料科学等领域的其他研究。
现代QSAR建模的核心目标是利用统计学和机器学习方法揭示分子的目标性质与其结构特征之间的经验关系。采用不同维度的分子表征算法,可以构建一维(1D)、二维(2D)、三维(3D)或更高维度的QSAR模型(图1)。分子的1D描述符包括分子量、原子种类数、氢键供/受体数、环数以及特定官能团数。分子的2D描述符,如形状、大小、杂化类型和连接方式,可以通过分子图(molecular graph)来提取。分子的3D描述符,如极性表面积或3D原子对,可以通过分子几何结构(molecular geometry)来计算。而对于更高维度的特征,如分子的4D描述符,可以通过分子的构象集合(conformation ensemble)来获得。
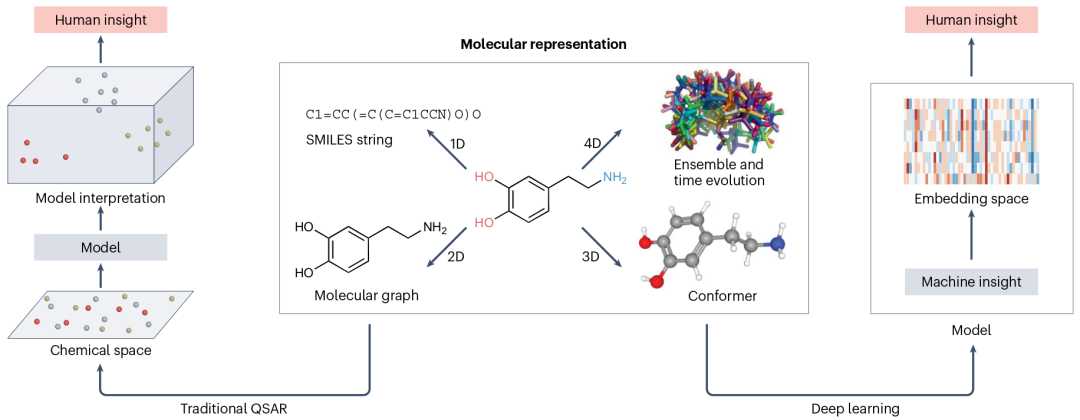
图1. QSAR和deep QSAR的异同
深度定量构效关系(Deep QSAR)
近年来,深度学习等人工智能(artificial Intelligence,AI)算法的发展、分子数据库的快速扩增,以及计算能力的提升,共同促进了深度定量构效关系(deep QSAR)建模的发展。与常见机器学习算法中的分子描述符不同(根据固定公式从化学结构中计算出来,且在训练过程中不发生变化),deep QSAR利用分子嵌入(molecular embedding)方法,从标准化学输入(如SMILES式或分子图)中创建的分子描述符(存在于深度学习算法创建的高维空间中的向量)(图1),可以随着模型训练的过程而改变,从而更好地用于预测目标性质。也就是说,deep QSAR通过在特定任务上训练深度学习模型,学习与分子或原子相对应的特征向量。因此,在模型训练过程中,分子嵌入和基于这种表征的模型训练成为模型优化过程中不可分割的部分。研究人员需根据经验或通过半自动方式(如进化算法、强化学习、元学习)选择合适的模型。Deep QSAR的优势在于其能处理多目标优化任务,并且在实际应用中,这一能力通过利用不同任务的数据来提高预测准确性。然而,在deep QSAR建模过程中,数据整理、模型适用范围以及独立模型验证仍然是关键的考量因素,并且随着数据量的增加,这些因素将面临更大的挑战。
Deep QSAR与生成模型
生成式药物分子设计
传统的QSAR模型可用于化合物库的虚拟筛选,以发现可直接购买的活性化合物。然而,药物化学的发展需要探索全新的化学实体,deep QSAR和分子生成模型的结合使得化合物的生成、打分和优化的整个过程能够实现一体化。
分子生成可分为基于化学反应规则(通过反应规则组装合成子)和基于深度学习的方法。后者首先学习训练集中分子的统计分布(即潜在空间,latent space),然后从中采样以生成新分子。由于训练过程中未引入详细的化学特征,使得分子生成过程难以用简单语言来描述。许多生成式药物设计方法采用深度神经网络(图2a-d)。其中最常见的方法是化学语言模型,利用文字的形式(如SMILES式)表示分子,学习分子间的内在语法以生成新分子。这类方法常用的算法包括循环神经网络(RNN)、变分自编码器(VAE)、生成对抗网络(GAN)和图神经网络(GNN)。还有一些模型采用了基于规则的分子生成与深度学习相融合的策略。
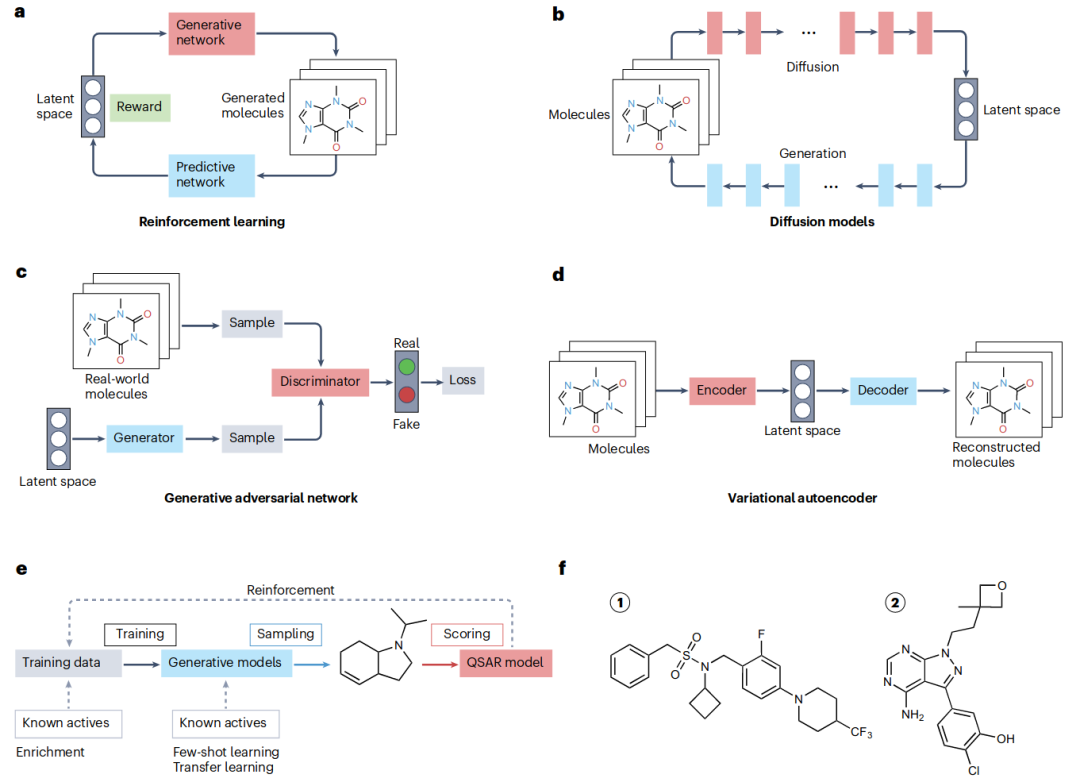
图2. 分子生成模型框架(a-e),生成式药物分子设计的案例(f)
在分子生成的过程中或之后,其目标活性可通过QSAR模型进行评价以供指导分子优化,该QSAR模型可以是独立于分子生成模型的外部评价器,也可以是整合在模型内置的打分函数(图2e)。部分研究还通过湿实验对模型生成分子进行验证,如化合物1(图2f)是一种新型的受体相关孤儿受体γ(RORγ)反向激动剂(IC50 370 nM),基于模型采样概率打分所得到。为了解决Deep QSAR模型在数据质量和模型适用性差时模型准确率降低的问题,David F. Nippa等人采用了基于模型集合的投票机制,发现了磷脂酰肌醇3-激酶γ(PI3Kγ)抑制剂化合物2(Ki 63 nM)(图2f)。
深度学习与合成路线设计
现有的生成式药物设计模型能够在几秒到几小时内为药物化学家提供一系列潜在的候选化合物。因此,目标化合物的合成往往成为进行候选化合物湿实验评价的限速步骤。合成路线的设计可以分为两类:逆向合成和正向合成。逆向合成从目标分子开始,寻找可能的反应前体和路径;而正向合成则是从已有的合成砌块开始逐步构建目标分子。
在逆向合成模型中,基于模板的策略(即反应转化规则),允许化学家提供模板或从反应数据库中自动提取模板,从而产生了LHASA、SYNTHIA、AiZynthFinder等逆合成分析软件。在无模板的逆合成策略中,深度学习方法直接从反应物和产物的化学结构出发,推断它们之间的关系,以预测逆合成路线和反应产物。例如,Jin等人利用含有注意力机制的图卷积神经网络来预测反应中心。基于自然语言处理,Sutskever等人开发了一种序列到序列的深度学习模型(以产物和反应物的SMILES作为输入),用于逆合成路线设计和反应产物预测。
在正向合成模型中新分子是逐步生成的,每步只有得分最高的产物才会进入下一步反应,最终所生成化合物的优劣可通过与目标分子的相似度来评价。这种策略可以用于合成产物的预测(如DOGS)、探索化学反应网络或生成虚拟化合物库。合成可行性(或其对立面,合成复杂性)是设计合成路线时的另一个重要评分指标,也可用于过滤虚拟化合物库。常用的打分程序包括SAscore(考虑了片段在PubChem中的出现频率以及分子的复杂程度)和SCScore(定义产物的复杂程度是否高于反应物)。除了合成可行性之外,化学反应的动力学和热力学特性在正向合成策略中也极为重要。这些信息可以通过反应浓缩图(condensed graphs of reaction, CGR)提取,并用于对双分子亲核取代反应、双分子消除反应、不同类型的环加成反应以及互变异构化反应的建模。
自动化分子设计
深度融合的分子生成模型、合成路线设计模型和deep QSAR模型,催生了自动化分子设计平台的诞生。这些平台能够自动创造出具有特定生物活性和理化性质的分子,并规划相应的合成路径。然而,随之而来的挑战包括如何在每个迭代循环(设计、合成、测试和分析)中高效处理大量数据,并实现多目标优化。目前,已有研究致力于利用自动化平台探索新的有机化学反应和加速药物设计过程。
Deep QSAR与基于结构的药物筛选
当目标蛋白的结构信息已知时,对大型分子库(超过100万个化合物)进行虚拟筛选是发现靶蛋白配体的有效策略。通常,这个过程包括对接(预测结合模式)和打分(评估结合模式)两个步骤。其中化合物的对接得分(docking score)应与其实验测定的结合能力相关。在传统的对接方法中,打分步骤只能在对接过程之后进行,这也导致了即使以现在的计算能力,也很难用这些方法对超大型化合物库(超过10亿个化合物)进行虚拟筛选。然而,随着近年来深度对接(deep docking)方法的发展,对超大型化合物库的筛选已成为可能。
渐进对接
早在2006年,研究人员已经开始在虚拟筛选中运用QSAR建模和主动学习(active learning)策略来预测分子的docking score,这一技术被命名为渐进对接(progressive docking)。在针对特定靶点的应用中,该技术首先对化合物库的部分分子进行筛选,获取其docking score,并以此作为QSAR建模的目标性质,建模过程中采用对计算资源需求较小的分子表征方式。最终,利用所得QSAR模型预测剩余分子的docking score,剔除低分分子,从而节约后续的计算资源。在Glide对接程序中,渐进对接算法得到了验证。线性QSAR模型的应用使Glide SP的虚拟筛选速度提升了2.6倍,同时保持了高达99%的苗头化合物(Hits)恢复度。后续研究还尝试引入非线性QSAR模型,例如NNscore。
深度对接
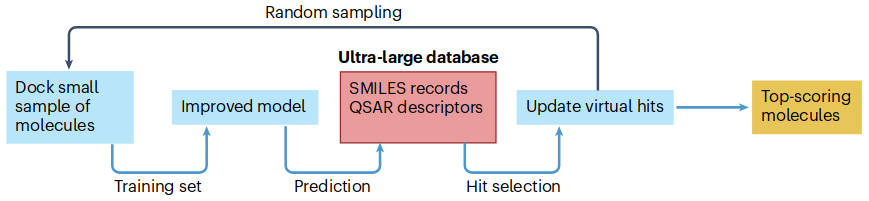
图3. 深度对接的工作流程
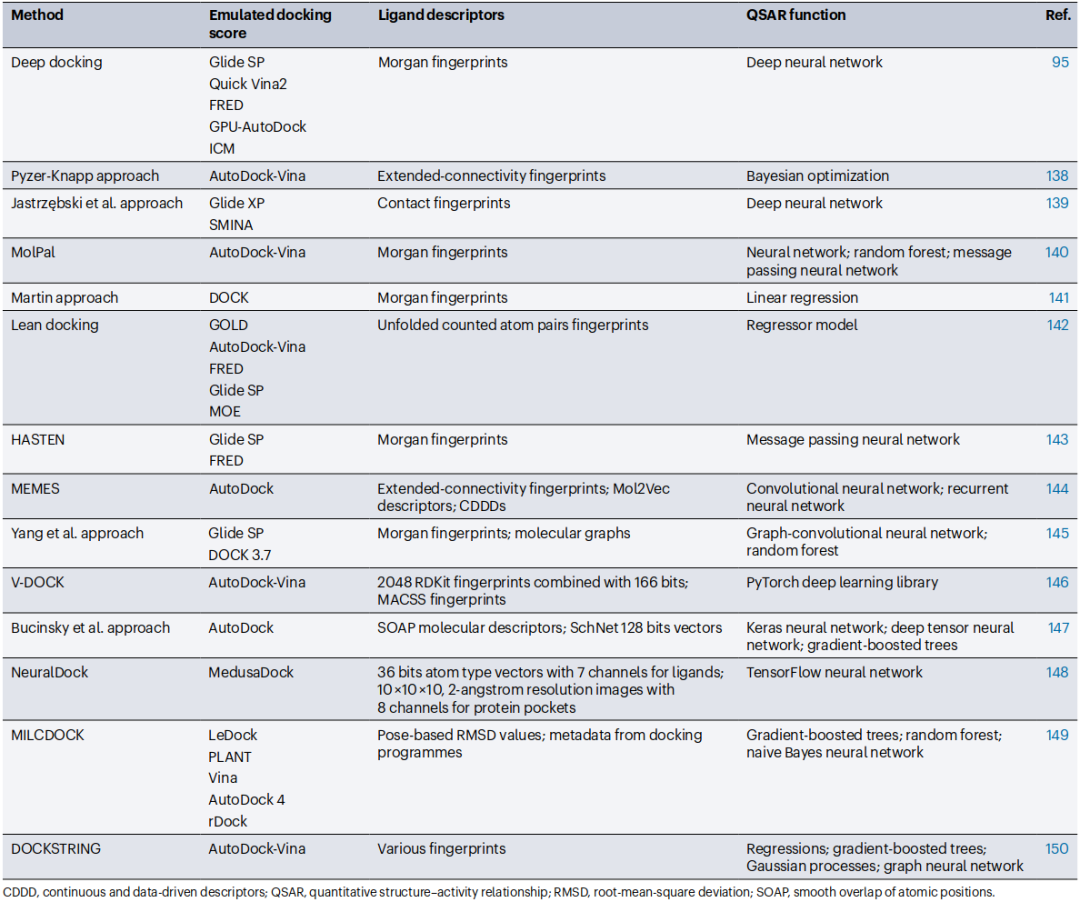
早期的这些研究能够在一定程度上提升对接速度(2-4倍),但仍然不足以应对数十亿级别分子库的筛选需求。因此,到了2020年,基于deep QSAR的深度对接(deep docking)方法应运而生(图3)。这种方法融合了主动学习和不依赖于靶蛋白的2D化学指纹,适用于对超大型化合物库进行虚拟筛选。例如,通过对ZINC15数据库中1.4亿个化合物的筛选,研究人员成功识别了1000个针对SARS-CoV-2 Mpro的苗头化合物(包含585个独特的分子骨架),这些化合物的后续生物活性测试证明了该策略的有效性。此后,众多深度对接算法相继被开发,并被广泛应用于超大型化合物库的虚拟筛选中(表1)。
表1. 利用机器学习预测docking score的虚拟筛选方法
共识对接
随着各种对接程序的出现,共识对接(consensus docking)(考虑多个程序的结果)得到了发展。Francesco Gentile及其团队开发了一种共识对接算法,该算法采用Glide、ICM、FRED、GPU-AutoDock和QuickVina2进行深度对接,筛选针对SARS-CoV-2 Mpro的化合物。利用250个GPU和640个CPU核心的计算资源,耗时大约90天,研究团队将最初的400亿分子库(来自ZINC15和Enamine REAL Space数据库)缩小了1500倍。结合这五种对接软件和两种筛选策略(分子结构聚类和药效团筛选),研究团队开发了28种共识对接策略,其中2种策略有计算机辅助药物设计(CADD)专家的参与。结果显示,在26种全自动共识对接策略中,最佳苗头化合物的命中率达到了13%,略低于CADD专家参与的2种策略(命中率分别为16%和23%)。
尽管深度对接显著加快了基于结构的药物发现过程,并提高了共识对接结果的准确性,但这些方法仍面临着一些需要解决的挑战。例如,深度对接虽然提升了速度,但其结果的准确性受限于当前对接软件打分函数的准确性。此外,如何选择合适的起始分子作为训练集用于docking score的生成,以及如何选择最合适的深度学习算法,都是目前尚未完全解决的问题。
多重药理学建模
大量的生物活性数据为开发多重药理学深度学习模型提供了丰富的资源。Li及其团队基于391种激酶与多种小分子抑制剂之间的约14万个活性数据,构建了一个激酶组多任务深度神经网络模型。这项工作使得研究人员能够描绘出一个包含靶向和脱靶效应的全面的激酶相互作用网络,该模型已被整合到KinomeX在线平台中。该平台能够仅基于化合物的2D结构(通过ECFP4指纹进行编码)来预测这些化合物对整个激酶组的多重药物学作用,为药物发现和再定位提供了强大的工具。
在大多数情况下,生物活性数据是有限的,而docking score的获得仅受计算资源的制约。此外,基于docking score训练深度学习模型时,并不需要关于“冷”靶点(即研究不足的靶点)的先验知识。例如,研究人员首先利用靶蛋白的同源蛋白抑制剂针对靶蛋白的活性口袋进行分子对接,并将得分高的分子列为训练集。然后,他们利用迁移学习的方法训练分子生成模型,同时将所有参与对接的分子用于训练docking score预测的深度学习模型。生成模型和预测模型通过强化学习模型相连,用于自动设计新分子并输出docking score(预测结果)高的分子。利用JAK1、JAK3和TYK2蛋白及其抑制剂的数据,该方法可用于寻找JAK2抑制剂。
未来展望
化学可及空间的扩增
近年来,点击化学、合成机器人和计算机辅助合成路线设计的发展,推动了化学数据库中分子数量的爆炸性增长。ZINC数据库的规模在16年间增长了5万倍(图4),同时,诸如CHEMriya、SAVI和Enamine REAL Space on-demand library等超大化合物库也相继问世。随着化学可及空间(accessible chemical space)的迅速扩增,如何高效筛选这些分子成为了新的挑战。例如,采用GigaDocking对嘌呤核苷酸酶和热休克蛋白90(Hsp90)进行的大规模虚拟筛选,动用了4.5万个CPU,在24小时内完成了对14亿个分子(来自Enamine REAL database)的筛选。尽管如此,这种庞大计算资源的投入限制了其普及性。因此,开发更高效、准确的虚拟筛选方法,并实现自动化的苗头化合物选择,特别是在集成AI算法的自动化合成实验室中,将成为未来研究的重要发展方向。
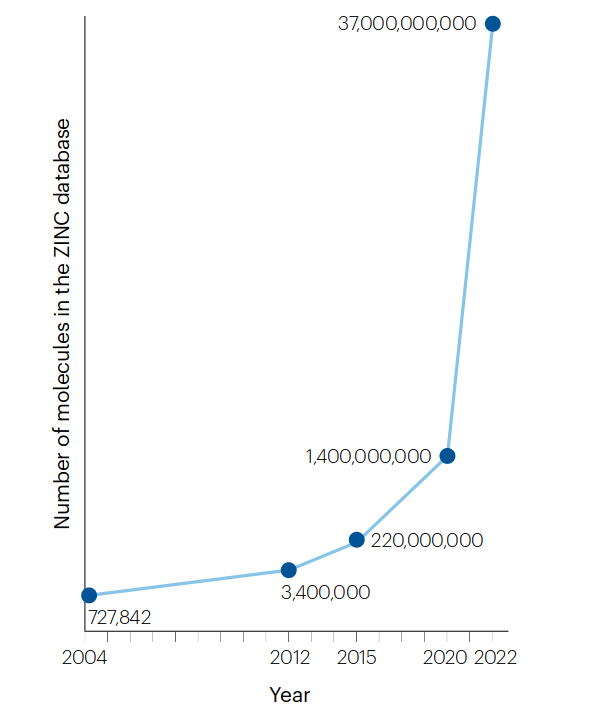
图4. ZINC数据库中的分子数量呈井喷式增长
深度学习加速量子力学计算
量子力学计算对计算资源的极高要求限制了其在QSAR建模和CADD领域的广泛应用。为了解决这个问题,计算化学领域长期致力于开发快速、精确且普适的量子力学近似方法,而深度学习技术的引入为这一领域带来了突破。
例如,原子神经网络势能(neural network potentials,NNPs)模型能够在保持密度泛函理论(density-functional theory,DFT)精度的同时,将分子能力以及其他量子力学性质的计算速度提升至DFT的106倍。然而,大多数NNPs在描述化学相互作用时存在局限性,这促成了更为通用的原子势能函数ANI-1的诞生。ANI-1成功地应用于预测包含环结构分子的稳定性、显著的构象变化以及蛋白-配体结合能(图5),在Schrödinger JACS标准集上的表现优于AMBER14SB/TIP3P力场。基于分子中原子(atoms in molecule,AIM)理论,电子密度分布函数将分子看成相互作用的原子的集合,AIMNet模型在ANI框架的基础上进行了改进。AIMNet模型学习到了新的原子特性,并在预测能量、原子部分电荷、原子体积等任务上展现了性能的提升。
总体而言,深度学习的融合不仅加快了量子力学近似方法的计算速度,还保持了其高精度,这将极大地推动分子对接、分子动力学模拟等CADD应用的发展。
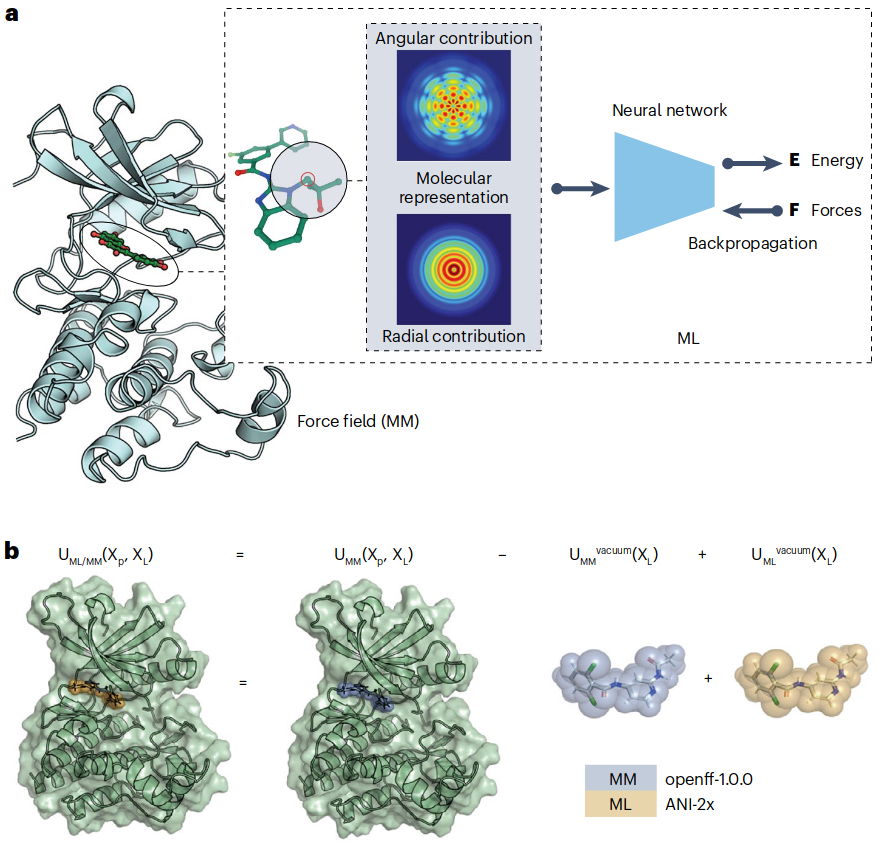
图5. 深度学习力场赋能小分子结合力预测
量子计算
量子计算(quantum computing)作为高性能计算领域的革命性突破,依托于量子力学的原理——量子比特(qubits)能够同时处于0和1的状态。这一特性使得量子计算机在处理庞大数据量时能够通过极少的操作实现显著加速,尤其在某些特定计算任务上展现出前所未有的速度优势。例如,在大型分子系统的薛定谔方程求解方面,量子计算机的应用预示着一系列革命性的突破:它不仅能够推动合成路线的设计,还能用于共价药物作用模式的建模、酶催化过程的模拟、量子化学表征方式的开发、以及靶标-配体相互作用的精确计算等多个领域。这一技术进步的将有效解决目前在处理超大型数据库和复杂量子力学计算方面所面临的挑战,极大地加速CADD领域的发展。
早期新药研发
尽管目前还没有药物基于深度学习算法和deep QSAR建模获得批准,但越来越多的证据显示,这些技术正在快速推动小分子药物的临床前研究。例如,Exscientia在2020年宣布了首个由人工智能设计的分子进入I期临床试验,临床前研究仅用了12个月时间。随后,Insilico Medicine在2022年报道,通过其Pharma.AI人工智能平台,从新靶点发现到分子设计再到进入一期临床研究,整个过程仅耗时30个月。这两家公司都在其AI平台整合了deep QSAR算法。这些最新的成功案例表明,deep QSAR技术已经进入了Gartner曲线(Gartner hype cycle)中的“生产力平台(plateau of productivity)”阶段(图6)。
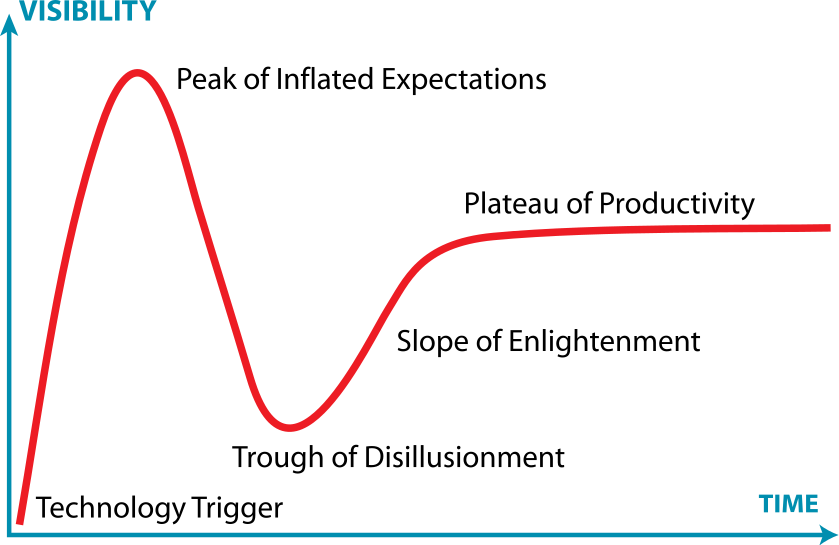
图6. Gartner曲线
随着deep QSAR方法的不断发展和应用,我们可以预期小分子药物候选物的发现将得到极大的加速,这将有助于人类更好地应对新的、无法预料的疾病风险,如COVID-19。
参考资料:
1. Tropsha, A.; Isayev, O.; Varnek, A.; Schneider, G.; Cherkasov, A. Integrating QSAR Modelling and Deep Learning in Drug Discovery: The Emergence of Deep QSAR. Nat. Rev. Drug Discov. 2023, 1–15. https://doi.org/10.1038/s41573-023-00832-0.
2. https://en.wikipedia.org/wiki/Gartner_hype_cycle
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-01-26,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号