ICML 2024 | 统一分子建模中的多尺度蛋白质语言模型

ICML 2024 | 统一分子建模中的多尺度蛋白质语言模型

DrugOne
发布于 2024-06-18 15:42:50
发布于 2024-06-18 15:42:50
今天为大家介绍的是来自北京大学Kangjie Zheng与南京大学Siyu Long等人发表的一篇论文。蛋白质语言模型在蛋白质工程领域展示了显著的潜力。然而,当前的蛋白质语言模型主要在残基层面操作,这限制了它们提供原子级别信息的能力,阻碍了在涉及蛋白质和小分子的应用中充分发挥蛋白质语言模型的潜力。在本文中,作者提出了ms-ESM(多尺度ESM),这是一种新的方法,能够实现多尺度统一分子建模。ms-ESM通过在多尺度Code-Switch蛋白质序列上进行预训练,并利用多尺度位置编码来捕捉残基和原子之间的关系,从而实现这一目标。实验结果表明,ms-ESM在蛋白质-分子任务中超越了以往的方法,充分展示了蛋白质语言模型的潜力。进一步研究表明,通过统一的分子建模,ms-ESM不仅获得了分子知识,还保留了对蛋白质的理解。

蛋白质语言模型(PLMs)在蛋白质工程领域展示了显著的潜力,能够在大规模蛋白质序列的预训练中捕捉到生化和共进化的知识。这使得它们在包括蛋白质结构预测、蛋白质适应性预测和蛋白质设计在内的各种领域取得了显著成就。然而,当前的PLMs主要在蛋白质残基(氨基酸)层面上操作,无法提供原子级别的信息。这限制了PLMs在涉及蛋白质和小分子的应用中的潜力。因此,外部的小分子模型必须纳入以解决这些应用。然而,蛋白质也是由原子组成的,仅在残基层面上建模蛋白质可能会导致低分辨率,无法捕捉到原子层面的信息。扩展PLMs以同时在残基和原子层面上操作,可以使其应用范围更广。
模型框架
概述
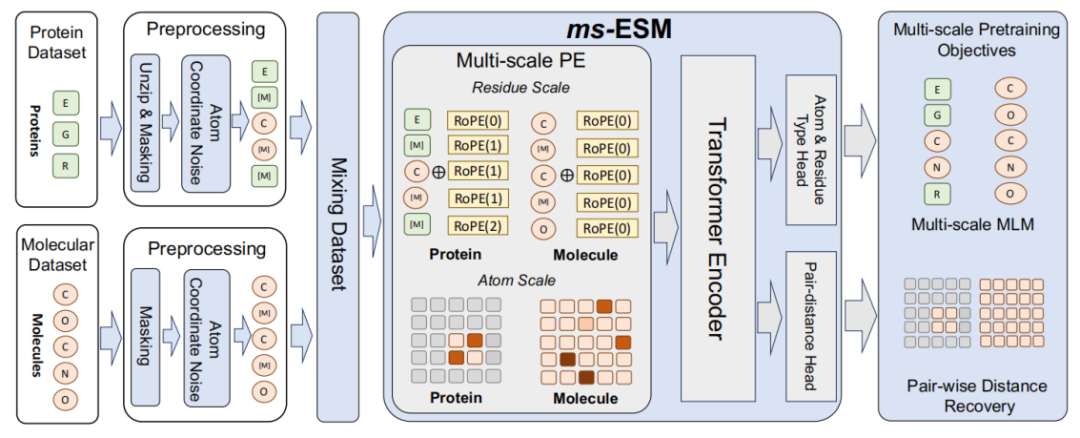
图1:多尺度预训练框架
作者首先概述了多尺度预训练模型,即ms-ESM(见图1)。对于数据集D中的每个数据 Xi,作者通过解压部分残基创建其Code-Switch序列,之后采用掩码语言建模(MLM)和成对距离恢复(PDR)作为预训练任务。为了处理序列中残基和原子共存的情况,作者提出了一种多尺度位置编码(MSPE)来描述复杂的位置关系。
多尺度预训练
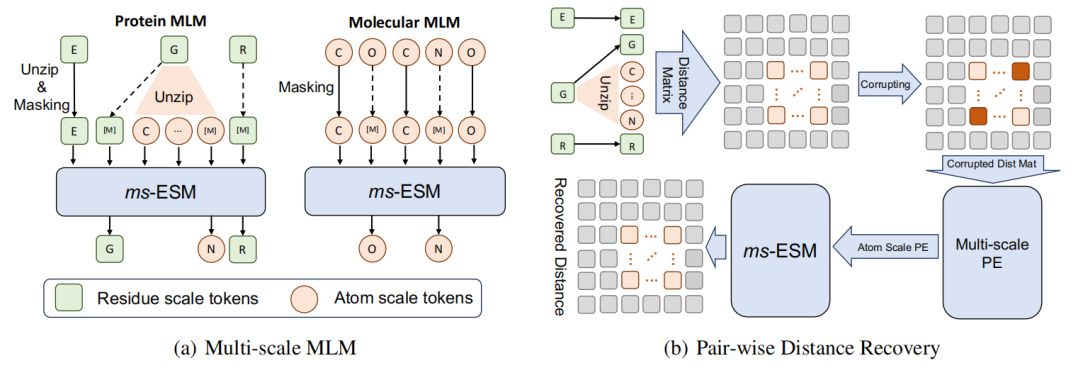
图2:多尺度掩蔽语言建模和成对距离恢复
如图2所示,创建Code-Switch蛋白质序列并在其上实现预训练任务(即MLM和PDR)的具体过程如下:
Code-Switch蛋白质序列:通过随机选择一组残基并插入其对应的原子到序列X,构建Code-Switch蛋白质序列。这一过程被视为一种解压操作。在实际建模中,可以解压多个残基。在解压过程中,赋予解压后的原子顺序。解压后的序列包含原始残基和解压后的原子信息。
掩码语言建模(MLM):在Code-Switch序列上随机掩盖部分原子或残基,然后要求模型利用周围的上下文预测原始的原子或残基。
成对距离恢复(PDR):使用添加噪声的原子作为模型输入,要求模型恢复这些原子之间的精确欧几里得距离。通过向坐标添加噪声并要求模型重建实际距离,ms-ESM可以学习到丰富的残基结构知识。
多尺度位置编码
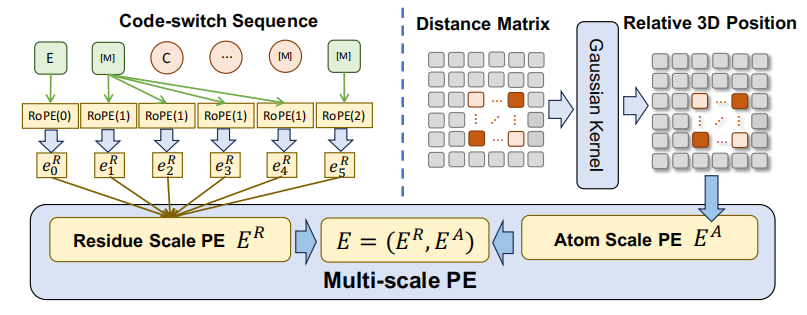
图3:多尺度位置编码框架
设计多尺度位置编码E来编码Code-Switch序列中的位置关系。E包含残基尺度位置编码ER和原子尺度位置编码EA。图3说明了作者多尺度位置编码的框架。
残基尺度位置编码(ER):使用旋转位置嵌入(RoPE)描述Code-Switch序列中残基之间的位置关系。当处理两个来自同一残基的原子时,ER不会引入任何模糊位置信息。
原子尺度位置编码(EA):为了描述原子之间的关系,直接使用欧几里得距离矩阵和高斯核来编码原子的3D位置。
将多尺度位置编码集成到Transformer中
ms-ESM的参数化略有不同于原始Transformer架构。具体来说,用残基尺度位置编码ER替换Transformer中的正弦编码。对于原子尺度位置编码EA,将其视为自注意力层的偏置项。通过对原始Transformer架构的轻微修改,ms-ESM能够同时处理残基和原子信息,成为各种下游任务的通用模型。
实验结果
表1:在酶-底物亲和力回归(ESAR)任务和酶-底物对分类(ESPC)任务上的性能比较
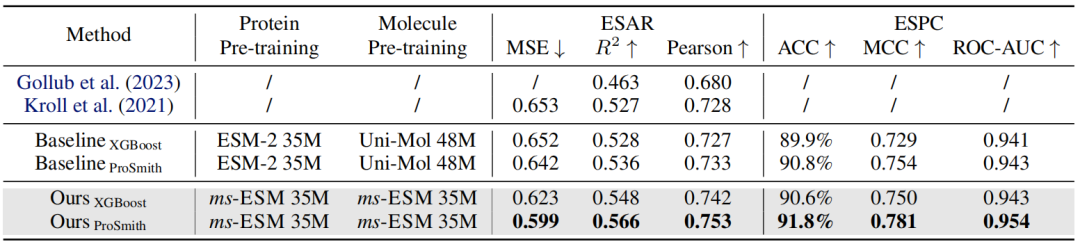
表2:药物-靶标亲和性回归任务的性能比较
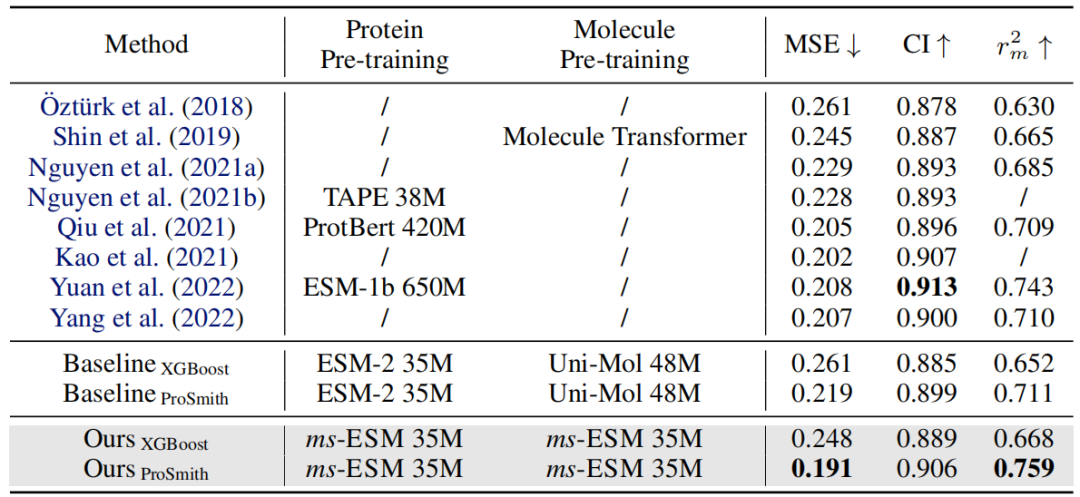
表1和表2展示了ms-ESM和基线模型在这三个任务上的实验结果。根据结果,作者可以总结出以下发现:
(i)ms-ESM在大多数指标上表现优于其他模型,并达到了最先进的水平。(ii)基于ms-ESM的微调策略,如ProSmith和XGBoost,表现一直优于结合两个独立预训练模型的版本。(iii)ms-ESM甚至可以超越基于更大预训练模型的方法。这些发现清楚地表明,ms-ESM在残基和原子层面上均能成功运作,并且在单个模型中预训练蛋白质和分子可以充分利用预训练技术在蛋白质-分子任务中的潜力。将两个独立的预训练模型融合在此类任务中可能是不理想的,且这个问题不能通过使用更大的预训练模型来解决。
表3:接触预测(CP)任务与二级结构预测(SSP)任务的性能比较
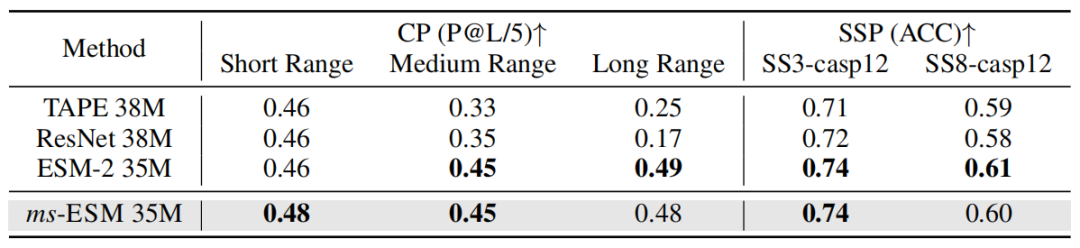
表3报告了接触预测和二级结构预测的结果。虽然ms-ESM可能不能在比较的方法中获得最好的性能,但它在二级结构预测和接触预测方面的性能与ESM-2相似。这表明ms-ESM并没有牺牲其对蛋白质的理解。幸运的是,ms-ESM可以通过用更大的ESM-2初始化其参数来提高对蛋白质的理解。
表4:消融实验结果
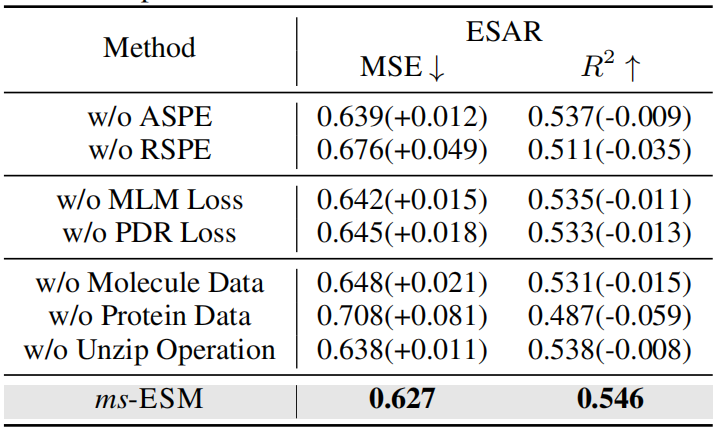
为了验证多尺度位置编码的有效性,作者在两种条件下进行了消融测试:一种是不使用原子尺度位置编码(ASPE),另一种是不使用残基尺度位置编码(RSPE)。所使用的任务是酶-底物亲和力回归。正如表4所示,当省略原子尺度位置编码或残基尺度位置编码时,模型的性能显著下降。这是因为在缺少位置编码的情况下,模型无法捕捉原子和残基的位置信息。这些结果证明了作者多尺度位置编码的有效性。
当省略掩码原子类型预测损失或成对距离恢复损失时,模型性能显著下降。值得注意的是,省略成对距离恢复损失导致的性能下降比省略掩码原子类型预测损失更为严重。这可能是因为没有成对距离恢复损失,ms-ESM无法在原子尺度上学习结构信息。这些结果表明,虽然原子类型和结构信息对于原子尺度的细节都很重要,但结构信息更为重要。
当排除分子数据或蛋白质数据时,模型性能显著下降,其中移除蛋白质数据导致的性能下降比移除分子数据更为显著。这表明当模型未使用蛋白质数据进行训练时,它会迅速失去与蛋白质相关的知识,导致整体性能显著下降。然而,即使没有分子数据,模型仍然可以通过解压操作获取原子尺度信息。因此,模型在没有分子数据的情况下表现优于没有蛋白质数据的情况。此外,当省略解压操作时,模型的性能显著下降。这些结果证实了解压操作的有效性。
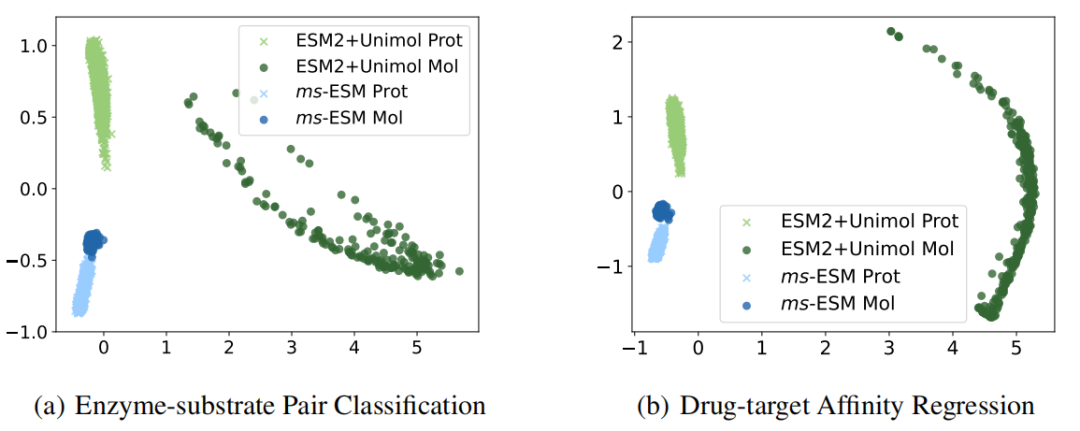
图4:ms-ESM与ESM-2+Uni-Mol学习到的表征可视化
为了更直观地展示ms-ESM学习到的蛋白质和小分子表示的高质量,作者在酶-底物对分类和药物-靶标亲和力回归任务中,对从ms-ESM和ESM-2+Uni-Mol提取的表征进行了可视化比较。具体来说,作者使用经过微调的模型(即表1和表2中的Baseline ProSmith和Ours ProSmith)来提取蛋白质和分子的表示,随后使用主成分分析(PCA)来可视化这些表征。
如图4所示,ms-ESM模型学习到的蛋白质和分子表示更加紧密地对齐。这表明ms-ESM模型能够创建一个更具凝聚力的语义表示,涵盖了蛋白质和分子数据,从而使ms-ESM在性能上优于两个独立的预训练模型。
结论
在本研究中,作者提出了一种多尺度蛋白质语言模型 ms-ESM,该模型通过在多尺度Code-Switch蛋白质序列上进行预训练,并使用多尺度位置编码来描述残基和原子之间的关系,从而实现了多尺度统一分子建模。实验结果表明,ms-ESM 在蛋白质-分子任务中表现优于以往的方法,并且在不牺牲对蛋白质理解的前提下,成功地将分子知识整合到蛋白质语言模型中。
编译|于洲
审稿|曾全晨
参考资料
Zheng K, Long S, Lu T, et al. Multi-Scale Protein Language Model for Unified Molecular Modeling[J]. bioRxiv, 2024: 2024.03. 04.583284.
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-06-11,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号