大可不必删除一个样品


交流群的小伙伴反馈说他在复现一个单细胞转录组数据集的数据挖掘文章:《 Comprehensive scRNA-seq analysis to identify new markers of M2 macrophages for predicting the prognosis of prostate cancer 》,对于作者“莫名其妙”的删除了一个样品的操作感觉“百思不得其解”:

“莫名其妙”的删除了一个样品
我下载了文献读了一下,发现正文里面压根就没有然后单细胞数据处理图表,完全就是最常规的预后模型的建立,基于tcga数据库的转录组测序表达量矩阵。单细胞内容都在附件里面,尤其诡异的是 文章里面提到了:Cluster analysis was carried out by Harmony and CCA ,感觉就是倚天屠龙都用上了,但是降维聚类分群后,仍然是每个病人的细胞分离很开,比如p4 泾渭分明 :
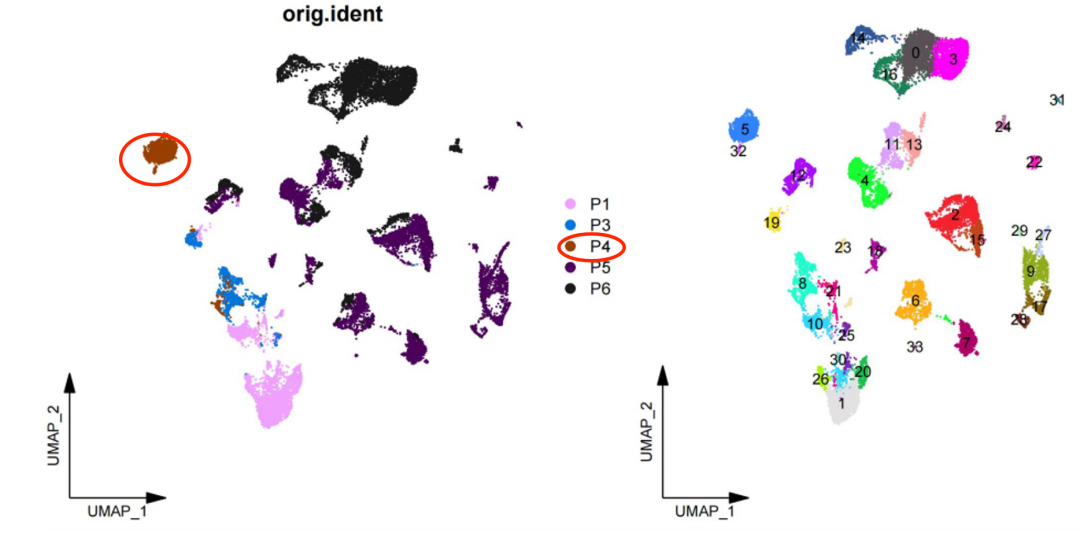
如果是按照文章里面的说法,那么p4也应该是被删除了。很明显是作者在做单细胞转录组数据处理的时候没搞懂Harmony的用法,如果是正常的Harmony后的降维聚类分群,然后给每个单细胞亚群一个生物学名字,会有如下所示的结果:
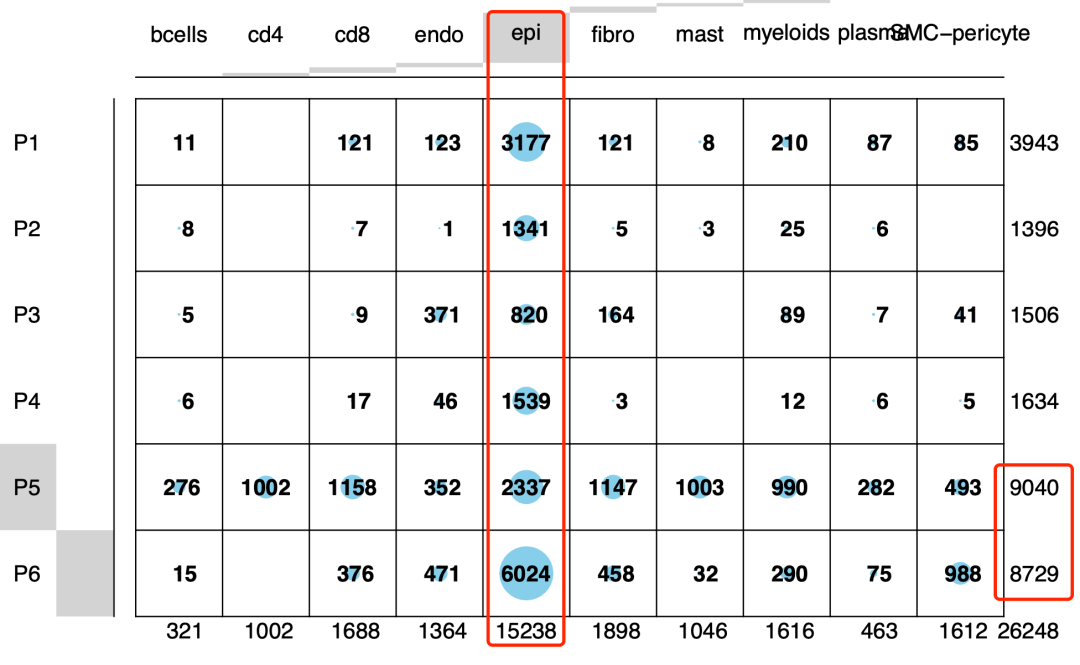
给每个单细胞亚群一个生物学名字
可以很明显的看到,如果不harmony那么p5和p6确实是会也有一些重叠,因为里面有免疫微环境细胞,同理p1和p3也是有一点点混合的细胞亚群,但是p2和p4都会比较特立独行,因为这两个样品的上皮细胞比例实在是太高了,而这个prostate cancer就是上皮细胞癌症,所以上皮细胞大概率上是会恶化的,而恶化后的肿瘤上皮细胞具有病人异质性,理论上每个病人都会跟其他病人格格不入的。
这就是为什么作者最开始会说因为P2跟其他病人格格不入所以删除了P2,实际上按照作者错误的做法,p4也可以同样的理由删除掉。因为作者压根就不知道的如何合理的使用harmony,如下所示可以看到左边的harmony成功的UMAP图里面就不会有p2或者p4的特立独行了!
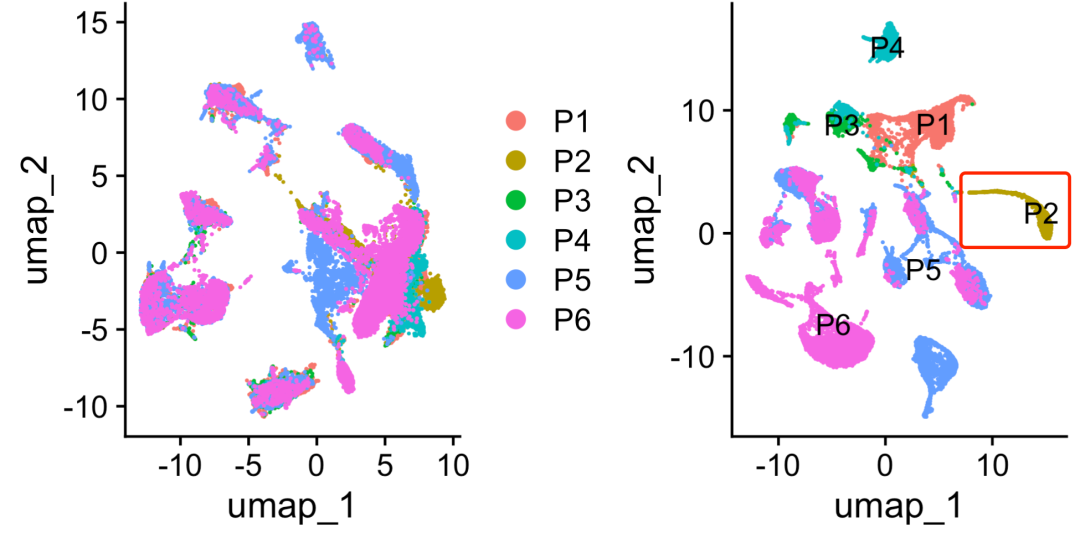
左边的harmony成功的UMAP图
成功的harmony之后就可以根据每个单细胞亚群特异性的基因去给生物学名字了,我给出来的示范如下所示:
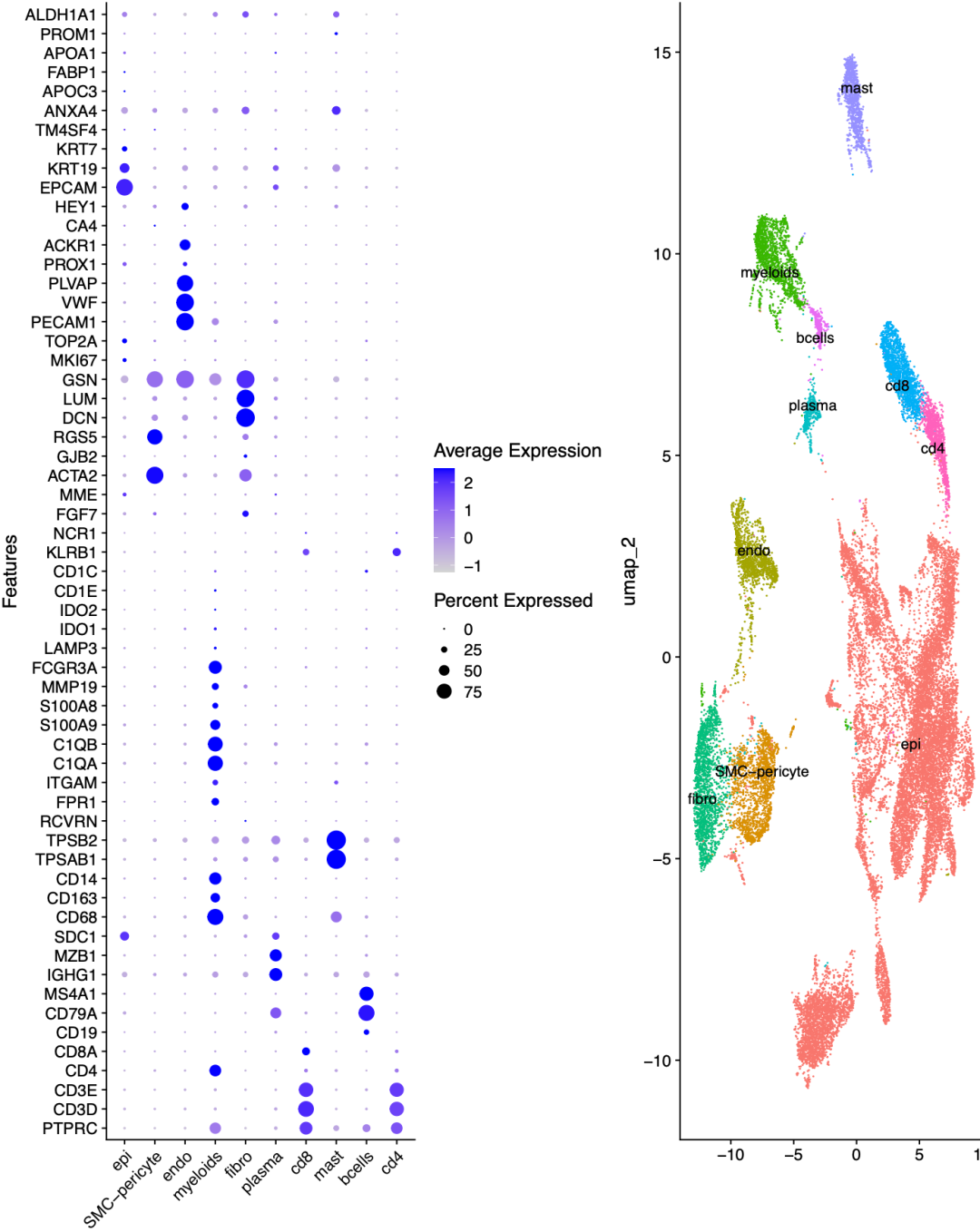
根据每个单细胞亚群特异性的基因去给生物学名字
这个时候的上皮细胞是多个病人被我们的harmony强行的混合在一起了,后续还有很多不同的数据处理流派来处理这个病人异质性!
seuratObj <- RunHarmony(input_sce, "orig.ident")
names(seuratObj@reductions)
seuratObj <- RunUMAP(seuratObj, dims = 1:15,
reduction = "harmony")
# p = DimPlot(seuratObj,reduction = "umap",label=T )
# ggsave(filename='umap-by-orig.ident-after-harmony',plot = p)
input_sce=seuratObj
input_sce <- FindNeighbors(input_sce, reduction = "harmony",
dims = 1:15)
source('scRNA_scripts/lib.R')
sce.all.int = readRDS('2-harmony/sce.all_int.rds')
sp='human'
colnames(sce.all.int@meta.data)
table(sce.all.int$RNA_snn_res.0.8)
seuratObj <- RunUMAP(sce.all.int, dims = 1:15 )
colnames(sce.all.int@meta.data)
DimPlot(sce.all.int, reduction = "umap", group.by = "orig.ident" )+
DimPlot(seuratObj, reduction = "umap" , group.by = "orig.ident",label = T)
其实这个RunHarmony大家都会使用,但是后面的RunUMAP函数需要基于前面的RunHarmony的结果,很多人就会做错了,这就是为什么很多人声称自己做了harmony的多个单细胞转录组样品的整合,但是没有效果。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-10-08,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号