NeurIPS | 柔性蛋白建模进入 3D 分子生成:FlexSBDD 的技术逻辑与局限

NeurIPS | 柔性蛋白建模进入 3D 分子生成:FlexSBDD 的技术逻辑与局限

DrugIntel
发布于 2026-07-13 16:21:52
发布于 2026-07-13 16:21:52

文献来源
论文题目: FlexSBDD: Structure-Based Drug Design with Flexible Protein Modeling 作者: Zaixi Zhang, Mengdi Wang, Qi Liu 机构: 中国科学技术大学、Princeton University 会议: NeurIPS 2024 代码链接: https://github.com/zaixizhang/FlexSBDD 论文类型: 方法论文,核心目标是在生成 3D 配体分子的同时,对蛋白结合口袋进行柔性构象更新。论文明确指出,将 FlexSBDD 定位为一个能够从 apo 蛋白结构出发,同时输出生成配体和更新后 holo 蛋白结构的生成模型。
导读
基于结构的药物设计中的 3D 分子生成通常把蛋白口袋视为静态环境:给定一个固定蛋白结构,模型在口袋中生成配体原子类型与坐标。但真实结合过程往往伴随诱导契合,尤其是侧链旋转、局部 loop 调整和隐性口袋暴露。FlexSBDD 的价值在于,它把配体生成和蛋白构象更新放进同一个 flow matching 生成框架中:蛋白以 Cα 坐标、骨架取向和侧链二面角表示,配体以原子类型和坐标表示,模型通过 E(3)-等变的标量-向量网络同时预测多种变量的流场。实验显示,FlexSBDD 在 CrossDocked 和 Binding MOAD 上取得更优 Vina 相关指标,并显著减少 steric clashes、增加氢键和疏水相互作用。它不是完全解决蛋白柔性的终点,但为生成式 SBDD 提供了一个重要方向:未来的模型不能只学会填口袋,还要学会让口袋参与设计。
为什么这篇论文值得关注?
生成式 SBDD 过去几年发展很快,TargetDiff、DecompDiff 等模型已经能在蛋白口袋内非自回归地生成 3D 配体分子。但这些方法大多隐含一个强假设:蛋白结构是固定的,配体必须适配一个预先给定的刚性口袋。
这个假设在计算上方便,却和真实结合过程存在差距。蛋白不是刚体。配体进入口袋后,局部残基可能旋转,loop 区域可能移动,侧链可能重新包装,甚至某些原本不可见的隐性口袋会被打开。论文在引言中直接指出,忽略蛋白柔性会带来三个问题:生成复合物容易出现不合理的蛋白构象和空间冲突;生成空间被已有 holo 结构限制;计算模拟与真实结合过程之间存在差距,可能增加实际药物发现中的假阳性。
FlexSBDD 的核心关注点正是这个长期存在但在生成式 SBDD 中处理不足的问题:能不能在生成小分子的同时,让蛋白口袋也发生合理的构象调整?
这使它区别于普通的 3D ligand generation,也区别于只做柔性 docking 的模型。它尝试处理的是更难的问题:输入一个初始化蛋白结构,通常可以理解为 apo 或近似 apo 状态,模型不仅要生成 ligand,还要预测蛋白从 ligand-free 到 ligand-bound 状态的局部构象变化。论文把这个目标写成条件生成分布:给定蛋白 P,生成更新后的蛋白 P′ 和配体 G,即学习 p({P′, G}|P)。
研究背景:刚性口袋为什么不够?
基于结构的药物设计依赖一个基本前提:如果知道靶蛋白的三维结构,就可以在结合位点中寻找或生成能够形成稳定相互作用的小分子。传统 docking、基于深度学习的打分函数、3D 分子生成模型,都围绕这个逻辑展开。
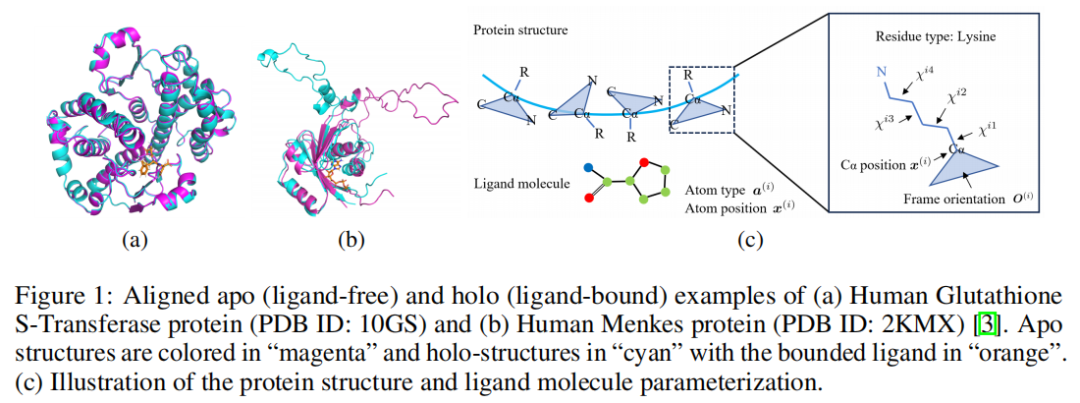
但真实世界中的结合过程不是把一个刚性钥匙插进一个刚性锁孔。诱导契合理论强调,配体结合会诱导蛋白发生构象变化,进而增强结合亲和力。论文中用 apo 与 holo 结构对齐示例说明,同一个蛋白在未结合和结合配体时可能存在明显结构差异。
在生成式 SBDD 里,刚性假设尤其容易带来问题:
第一,模型只能在固定口袋边界内生成分子。如果口袋中某个侧链在真实结合时会旋转让位,但模型训练和采样时固定该侧链,那么本来合理的分子可能被误判为冲突,或者模型干脆不探索这一类分子。
第二,模型可能过度拟合已有 holo 构象。已有复合物结构通常只展示某一个配体诱导出的结合状态,而不是完整构象空间。如果模型只学习固定 holo 口袋中的原子分布,它生成的分子往往更像是在复刻已有口袋填充模式。
第三,空间冲突会被放大。生成模型在口袋中直接放置原子,如果蛋白侧链不能适应配体,配体与蛋白原子之间容易出现低于范德华半径之和的接触距离。论文后续用 PoseCheck 分析 steric clashes,也正是为了评价这一点。
以往方法
已有 SBDD 生成方法大致可以分为三类。
第一类是基于 3D 网格或密度的生成方法。 例如 LiGAN 把蛋白-配体复合物表示成三维原子密度,利用条件 VAE 生成 ligand density,再通过 atom fitting 和 bond inference 得到分子。优点是形式直观,适合 CNN 处理三维空间;缺点是分辨率受限,旋转等变性处理不自然,且对原子级几何细节和柔性构象建模能力有限。
第二类是自回归生成方法。 例如 Pocket2Mol 逐个原子生成 ligand,预测下一个原子类型、坐标和键类型;片段式方法则逐步添加分子片段。它们能够利用局部上下文,生成过程也比较符合化学构建直觉,但容易出现误差累积,而且生成速度受限。
第三类是扩散式或非自回归生成方法。 TargetDiff、DecompDiff 等模型把蛋白-配体复合物表示为 3D 点集,对配体坐标和原子类型进行加噪、去噪,从而一次性或并行地生成分子。它们提升了生成质量和采样灵活性,但通常仍然把蛋白作为固定条件,蛋白只是背景环境,不参与构象更新。论文也明确指出,尽管 diffusion-based SBDD 已取得显著进展,蛋白柔性仍然常被忽略。
此外,柔性蛋白建模本身并不是没人研究。MD 可以模拟蛋白动力学,但计算成本高;DynamicBind、FlexPose、SBAlign、NeuralPlexer、DiffDock-Pocket 等方法已在蛋白-配体 docking 或复合物结构预测中考虑柔性。然而,从柔性 docking 扩展到 de novo ligand generation 并不直接,因为生成任务还要同时处理分子拓扑、原子类型、坐标分布、结合相互作用和化学合理性。论文把这视为一个尚未充分解决的问题。
FlexSBDD 的核心思想
FlexSBDD 的中心思想可以概括为一句话:
把蛋白构象变化和 3D 配体生成统一到同一个条件 flow matching 过程里,让模型从初始化复合物 C0 连续变换到目标 holo 复合物 C1。
这里的 C 代表 protein-ligand complex,由蛋白 P 和配体 G 组成。传统 SBDD 生成模型通常固定 P,只生成 G;FlexSBDD 则希望生成 {P′, G}。P′ 是经过 ligand-induced adjustment 后的蛋白结构,G 是新生成的 ligand。论文图 2 展示了这一流程:从 apo 蛋白结构和初始化 ligand 开始,每一个时间步都更新复合物 Ct,最终在 t=1 得到 holo-like protein-ligand structure。
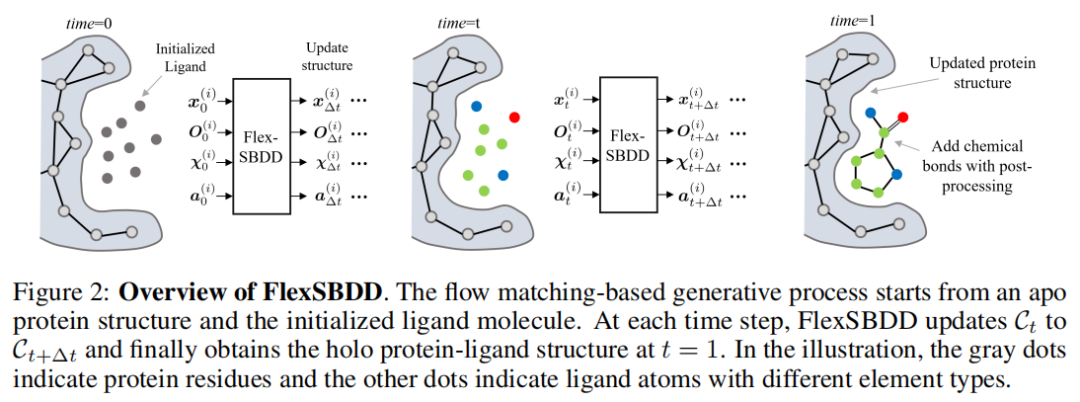
为什么选择 flow matching?因为普通扩散模型通常需要较多采样步数,在蛋白-配体柔性复合物这样高维系统中计算成本较高。Flow matching 直接学习从先验分布到数据分布的速度场,采样时通过 ODE 积分推进状态,可以用较少步数完成生成。论文默认使用 20 个 flow matching integration steps,而不是上千步扩散采样。
为什么不是全原子自由移动?因为蛋白原子数量大,自由度太高。FlexSBDD 采用一个折中方案:对蛋白残基使用紧凑的结构参数化,只建模 Cα 坐标、骨架 frame orientation 和最多 4 个侧链二面角。这样既保留了关键柔性自由度,又显著降低了建模复杂度。
5. 方法细节:模型到底是怎么做的?
5.1 任务定义与输入输出
FlexSBDD 的输入是初始化蛋白结构,可以是 apo conformation,也可以是近似 apo 的结构。模型还会初始化一个 ligand:先从参考数据集中采样 ligand 原子数,原子类型初始化为均匀分布,原子坐标在蛋白口袋内部用高斯分布初始化。
模型输出包括两部分:
- 1. 生成的 3D ligand:原子类型和三维坐标;
- 2. 更新后的蛋白结构:Cα 坐标、骨架取向和侧链二面角更新后重建出的 holo-like 构象。
因此,它不是单纯做 ligand generation,而是生成一个动态调整后的 protein-ligand complex。
5.2 蛋白如何表示:Cα + frame orientation + side-chain torsion
FlexSBDD 没有把蛋白每个原子都作为独立可移动点来建模,而是以残基为节点。每个氨基酸残基被表示为三个部分:
第一,Cα 坐标 x。 Cα 给出残基在三维空间中的位置,是蛋白 backbone 结构的核心锚点。
第二,骨架 frame orientation O。 O 属于 SO(3),表示残基局部骨架坐标系的方向。论文认为 backbone atoms 的位置可以由 Cα 坐标和 frame orientation 决定,因为 N、C、O 等骨架原子相对于 Cα 的理想局部坐标是已知的。
第三,侧链二面角 χ。 每个残基最多有 4 个侧链 torsion angles,取值范围在 [0, 2π)。侧链构象可以通过这些二面角和相对固定的键长、键角重建。论文把所有侧链二面角看作 hypertorus 上的变量,因为角度具有周期性,0 和 2π 表示同一个方向。
这个表示方式的优点是高效:一个残基对应一个节点,而不是十几个全原子节点。它也抓住了结合口袋中最关键的柔性来源之一:侧链旋转。缺点也很清楚:残基类型固定,不做蛋白序列设计;键长键角大多按理想值处理;更大尺度的构象变化、结构域运动、水分子、辅因子、质子化状态等并没有被完整纳入。
5.3 配体如何表示:原子类型 + 三维坐标
配体被表示为一组原子 G = {a, x}。其中 a 是原子类型概率向量,x 是原子坐标。也就是说,模型直接生成 ligand atom types 和 atom coordinates。
值得注意的是,论文核心建模对象是原子类型与坐标,而不是显式联合生成完整化学图的所有键类型。论文在评估中分析了生成分子的键长和键角分布,说明模型输出后可以构建分子并评估局部几何合理性;但从方法表述看,键连接并不是 FlexSBDD flow matching 的主要生成变量。
5.4 复合物图:蛋白残基和 ligand 原子共同作为节点
FlexSBDD 把 protein pocket-ligand complex 构造成 KNN 图。节点包括两类:蛋白残基节点和 ligand 原子节点。每个节点连接到最近的 k 个邻居,论文默认 k=8。
节点和边都有标量特征与向量特征:
标量特征主要承载化学身份信息,例如残基类型、原子类型概率、时间步嵌入等。时间步 t 用 sinusoidal embedding 表示,并拼接到节点标量特征中。
向量特征承载方向与几何信息。蛋白节点的向量特征来自骨架原子和侧链原子之间的欧氏向量组合,例如 C、CA、N、CB 之间的方向向量,以及侧链二面角相关原子之间的方向向量;ligand 节点的向量特征是 ligand 原子指向 ligand 几何中心的向量。边的标量特征使用 RBF 距离编码,边的向量特征使用相对坐标。
这个设计的意义在于:模型不只是知道两个节点距离多远,还能知道它们在三维空间中的相对方向。对需要移动原子、旋转侧链、更新骨架 frame 的任务来说,向量特征比单纯标量消息更合适。
5.5 Flow matching:模型学习的是从初始结构到 holo 结构的速度场
FlexSBDD 的生成过程不是传统意义上的逐步加噪再去噪,而是学习一个从 C0 到 C1 的连续流。C0 是初始化状态,包含 apo 蛋白和随机初始化 ligand;C1 是数据集中的 holo protein-ligand complex。模型学习在任意时间 t 下,当前状态 Ct 应该沿哪个方向移动,才能最终到达目标复合物。
不同变量处在不同数学空间中,因此 FlexSBDD 对它们分别定义 flow:
坐标变量 x 位于 R3。 包括蛋白 Cα 坐标和 ligand 原子坐标。它们采用线性插值,从 x0 移动到 x1。模型预测坐标速度场,训练时最小化预测速度与真实位移方向之间的差异。
骨架取向 O 位于 SO(3)。 旋转不能简单线性相加,因此论文使用 SO(3) 上的测地路径,通过 exponential map 和 logarithmic map 描述从初始取向到目标取向的旋转流。模型学习的是当前取向到目标取向的切空间方向。
侧链二面角 χ 位于 torus。 由于角度具有周期性,论文用 reg 函数把角度差规整到 [-π, π] 范围,避免把 359° 到 1° 错当成 358° 的大旋转。模型学习侧链角度的周期性流场。
ligand 原子类型 a 位于概率单纯形。 原子类型从均匀分布 a0 逐步流向 one-hot 的真实原子类型 a1。训练中使用交叉熵约束预测后的原子类型概率接近真实类型。
整体损失是四部分加权和:atom type loss、coordinate loss、orientation loss、side-chain torsion loss。默认权重为 2.0、1.0、1.0、1.0。训练使用 Adam,学习率 0.001,batch size 4,训练 500k iterations,单张 A100 约 36 小时。
5.6 网络结构:E(3)-等变的标量-向量双通道模型
FlexSBDD 的神经网络是 E(3)-equivariant network,并使用 scalar-vector dual representation。这里的 E(3)-equivariance 意味着,如果输入复合物整体旋转或平移,模型输出的坐标更新也会以一致方式旋转或平移,不会依赖任意坐标系。
网络包含 encoder 和 decoder。
Encoder 负责在 KNN 图上传递信息。每一层通过注意力机制计算邻居消息,并用 GVL、GVP、GVNorm、GVGate 等几何向量模块更新节点和边特征。GVL 的关键思想是让向量特征通过范数进入标量通道,同时让标量特征通过门控调节向量通道;GVP 在此基础上加入非线性变换。GVNorm 用于稳定标量和向量特征分布,GVGate 通过门控融合上一层特征和新消息。
Decoder 在编码后的特征基础上更新结构变量。它不仅更新节点特征,还显式更新坐标:一部分坐标变化来自节点自身的向量输出,另一部分来自邻居相对方向加权求和。最后,decoder 用 MLP 从最后一层表示预测 ligand 原子类型、残基侧链二面角和骨架 orientation。为了高效表示三维旋转,orientation 通过单位四元数预测,再转换为旋转矩阵。
这套结构背后的技术逻辑很清楚:蛋白柔性建模需要方向感,尤其是侧链旋转和局部骨架调整;单纯标量 GNN 很难直接表达三维方向更新,而标量-向量双通道网络更适合把化学身份和空间方向耦合起来。
5.7 数据增强:用 ApoBind + relax/repack 扩充 apo-holo 学习信号
柔性蛋白生成最大的难点之一是缺少成对的 apo-holo 数据。FlexSBDD 使用 ApoBind 提供的 apo conformations 与训练集 holo 结构建立配对,同时引入合成 apo 数据增强:先从 holo 蛋白中移除 ligand,再进行 OpenMM relaxation 和 Rosetta side-chain repacking。每个 holo structure 生成 9 个额外结构,包括 3 个仅侧链 repacking、3 个 relaxation + repacking、3 个额外加入最多 30° 侧链角扰动的结构。训练时,从对应 apo pool 中随机采样 C0,再和 holo C1 插值得到 Ct。
这个策略很实际。它并没有真正解决 apo-holo 数据稀缺问题,但增加了模型看到不同初始口袋构象的机会,使模型不只从单一 apo 状态学习到 holo 状态。
5.8 推理流程:从 apo 到生成复合物
实际生成时,流程可以概括为:
- 1. 输入目标蛋白结构,作为初始化蛋白 P0。
- 2. 根据参考分布采样 ligand 原子数。
- 3. 初始化 ligand 原子类型为均匀分布,坐标放在口袋中的高斯分布区域。
- 4. 构建 protein-ligand KNN 图,加入时间步嵌入。
- 5. 从 t=0 到 t=1 用 Euler solver 积分 ODE,每一步更新坐标、骨架取向、侧链二面角和 ligand 原子类型。
- 6. 最终得到 ligand 3D 结构和更新后的蛋白 holo-like structure。
这也是 FlexSBDD 与传统生成模型最关键的区别:生成过程中蛋白结构不是静态背景,而是随 ligand 一起被更新。
6. 实验设计与关键结果
6.1 数据集与对比方法
论文使用两个常见 SBDD benchmark:
CrossDocked:原始包含 2250 万 protein-molecule pairs,论文沿用既有预处理,只保留 docking pose 与真实 pose RMSD < 1 Å 的高质量样本,并采用 30% sequence identity split,得到 100,000 个训练对和 100 个测试蛋白。
Binding MOAD:包含约 41k experimentally determined protein-ligand complexes,按 enzyme commission number 过滤和划分,得到 40k 训练样本、100 验证样本和 100 测试样本。论文把这些复合物结构视为 holo structure,对应 apo structure 来自 ApoBind 和生成的 apo augmentation。
对比方法包括 LiGAN、AR、Pocket2Mol、TargetDiff、DecompDiff,覆盖了 3D CNN/VAE、自回归生成和扩散式生成方法。
6.2 评价指标:不能只看 Vina,还要看物理合理性
论文从三个维度评估:
第一,亲和力相关指标。 包括 Vina Score、Vina Min、Vina Dock 和 High Affinity。Vina Score 直接对生成构象评分;Vina Min 先局部结构优化再评分;Vina Dock 会额外重新 docking,用来估计可达到的较优 docking affinity;High Affinity 表示生成分子比参考分子得分更好的比例。
第二,药物相关性质。 包括 QED、SA 和 Diversity。QED 是类药性指标,SA 代表合成可及性,Diversity 计算同一口袋生成分子之间的平均不相似性。
第三,结构与相互作用合理性。 包括键长、键角分布的 Jensen-Shannon divergence,以及 PoseCheck 统计的 steric clashes、hydrogen bonds 和 hydrophobic interactions。论文附录中说明,clash 的判定基于蛋白原子与 ligand 原子距离是否低于范德华半径之和,并允许 0.5 Å tolerance。
这个评价体系比单纯看 docking score 更合理,因为生成式 SBDD 很容易出现 score 看起来不错但 pose 不物理的问题。
6.3 CrossDocked 主结果:更强的 Vina 指标和更少冲突
在 CrossDocked 上,FlexSBDD 的 Avg. Vina Dock 为 -9.12,Med. Vina Dock 为 -9.25;相比 DecompDiff 的 -8.39 和 -8.43 有明显提升。High Affinity 也从 DecompDiff 的 64.4% / 71.0% 提升到 FlexSBDD 的 78.5% / 84.2%。同时,FlexSBDD 的 QED 为 0.58 / 0.59,SA 为 0.69 / 0.73,Diversity 为 0.76 / 0.75,说明它并非只通过牺牲基础分子性质换取 docking score。
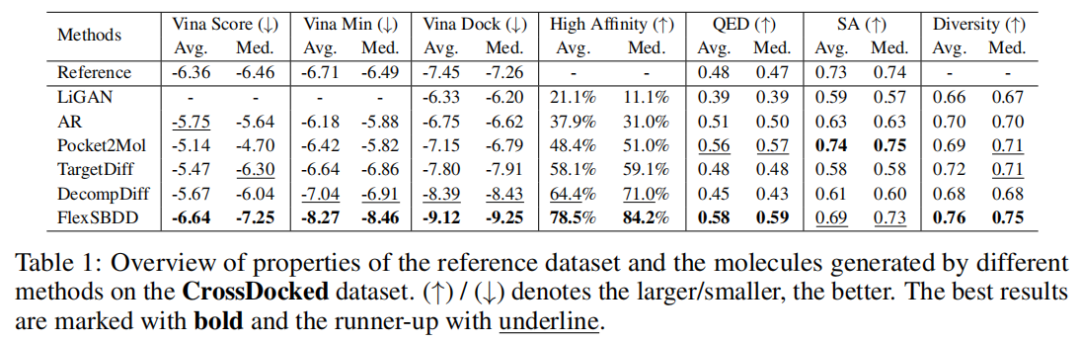
更值得关注的是相互作用分析。DecompDiff 的平均 steric clashes 为 6.43,而 FlexSBDD 降到 1.39;HB acceptors 从 1.18 提升到 1.96。这与论文主张一致:让蛋白参与柔性调整,有助于减少不合理空间冲突,并形成更多有利非共价相互作用。
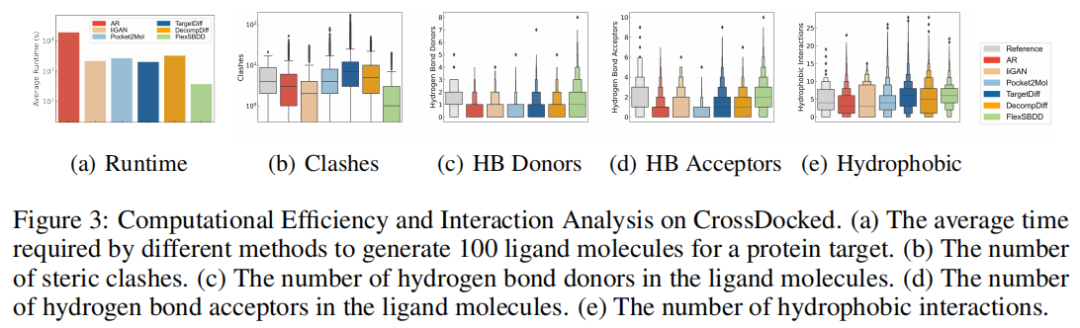
6.4 Binding MOAD:在实验复合物数据上仍有优势
在 Binding MOAD 上,FlexSBDD 的 Avg. Vina Dock 为 -9.38,Med. Vina Dock 为 -9.54;DecompDiff 分别为 -8.46 和 -8.51。High Affinity 从 DecompDiff 的 56.4% / 58.3% 提升到 74.5% / 76.9%。FlexSBDD 的 QED 为 0.63 / 0.64,SA 为 0.42 / 0.41,Diversity 为 0.73 / 0.74。
Binding MOAD 更接近实验复合物结构数据,因此这个结果支持了 FlexSBDD 不只是适配 CrossDocked 这类 cross-docking 数据,也能在实验结构集合上保持较好表现。不过需要注意,这仍然是 retrospective benchmark,不等同于真实项目中的 prospective hit discovery。
6.5 结构合理性:键长、键角、侧链预测与蛋白结构有效性
论文对生成 ligand 的键长和键角分布做了 JSD 分析。FlexSBDD 在多种键长类型上达到更低或接近最低的 JSD,例如 C=C、C−N、C−O、C=O、芳香 C:C 和 C:N 等,说明生成分子的局部几何分布更接近参考分子。
对蛋白结构,论文使用 scTM 评估更新后蛋白结构的结构有效性。FlexSBDD 更新结构的平均 scTM 为 0.964,接近数据集中原始结构的 0.975。侧链角度方面,FlexSBDD 在 χ1、χ2、χ3、χ4 上的 MAE 分别为 12.95、17.80、32.18、46.71,优于 NeuralPlexer 的 15.40、18.77、44.83、50.24。
这些证据支持一个较重要结论:FlexSBDD 并不是任意扭曲蛋白以迁就 ligand,而是在一定程度上保持了蛋白结构合理性。
6.6 消融实验:侧链柔性比骨架柔性更关键
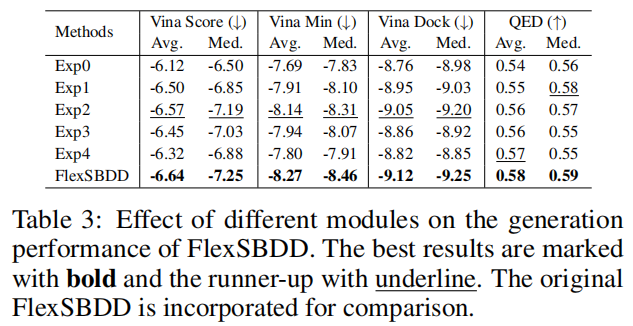
论文做了 5 组消融:
- • Exp0:移除数据增强;
- • Exp1:用只有标量特征的 EGNN 替换几何向量模块;
- • Exp2:不更新蛋白 backbone;
- • Exp3:不更新 side-chain torsion angles;
- • Exp4:固定整个蛋白结构。
完整 FlexSBDD 的 Avg. Vina Dock 为 -9.12。去掉 sidechain 更新后,Exp3 降到 -8.86;去掉 backbone 更新后,Exp2 为 -9.05;固定整个蛋白后,Exp4 为 -8.82。这说明侧链柔性对性能贡献尤其明显,也符合结合口袋中侧链重排常常直接决定相互作用和冲突消除的直觉。
附录中的相互作用消融进一步支持这一点:完整 FlexSBDD 的 steric clashes 最低,为 1.39,HB acceptors 为 1.96,hydrophobic interactions 为 6.12,均优于固定蛋白或去掉侧链柔性的变体。
6.7 KRASG12C 隐性口袋案例:有启发,但还不是实验验证
论文用 KRASG12C 做案例研究。作者以 ARS-1620 结合模式对应的结构作为 apo-like 输入,生成 ligand 后观察到 His95 侧链旋转,与文献中 AMG 510 相关结构观察一致,并形成新的 subpocket。论文认为这说明 FlexSBDD 具有探索 cryptic pockets 的潜力。
这个案例很有启发,因为隐性口袋正是蛋白柔性对药物发现最有价值的场景之一。但它仍然属于 retrospective structural case study。模型是否能在完全未知靶点上预测真正可成药的 cryptic pocket,还需要更多 prospective 设计、合成和实验验证。
启发
第一,生成式 SBDD 的下一步不只是更强的 ligand prior,而是更真实的 receptor response。 过去很多模型把口袋看作条件输入,配体看作生成对象。FlexSBDD 提醒我们,结构基础设计的对象其实是复合物,而不是孤立分子。
第二,蛋白柔性可以用分层自由度处理,而不必一开始就全原子 MD 化。 Cα 坐标、frame orientation、side-chain torsion 是一个实用折中。它不像全原子动力学那样昂贵,也比刚性口袋更接近结合过程。
第三,Flow Matching 适合处理多模态结构变量。 FlexSBDD 同时处理 R3 坐标、SO(3) 旋转、torus 角度和离散原子类型概率。这个思路对后续分子生成、蛋白设计、复合物建模都有启发意义。
第四,物理合理性评价正在变得和 docking score 同样重要。 论文不仅报告 Vina,还分析 clashes、氢键、疏水相互作用、键长键角分布和 sidechain MAE。这一点非常值得后续 SBDD 生成模型借鉴。
局限性
柔性仍然是有限柔性
FlexSBDD 建模了 Cα、骨架取向和侧链二面角,但没有完整模拟蛋白构象 ensemble。大尺度结构域运动、loop 长程重排、多构象选择、allosteric coupling、水分子网络、金属离子、质子化状态等仍然没有系统纳入。
这意味着它更适合处理局部 pocket adjustment 和 side-chain repacking,而不是所有蛋白柔性问题。
数据增强可能保留 holo 偏置
合成 apo 结构来自移除 holo ligand 后的 relaxation 和 repacking。虽然这些结构不含 ligand,形式上对 baselines 公平,但它们的初始几何仍可能保留 holo 口袋痕迹。这会让模型更容易学习从近 holo 状态回到 holo 状态,而不是从真实 apo ensemble 预测真实 ligand-induced transition。
Vina 和 Glide 仍然不是实验亲和力
论文补充使用 GlideSP min-in-place score 评估,FlexSBDD 得到 -7.55,优于 DecompDiff 的 -7.09 和其他方法。 但 docking score 本质上仍是近似评分函数,不能代表真实结合自由能。生成模型可能学会优化评分函数偏好,而不一定提高实验命中率。
缺少湿实验验证
KRASG12C 案例展示了结构上的一致性,但没有合成新分子并进行生化或细胞实验验证。对于 drug discovery 任务,最终证据仍然是实验活性、选择性、构效关系和可优化性。
分子化学有效性仍需更强约束
FlexSBDD 主要生成 atom types 和 coordinates,局部键长键角分布表现不错,但真实可合成性、反应可达性、代谢稳定性、毒性风险、PAINS、聚集体、共价反应性等都不在核心训练目标中。SA 和 QED 只能作为粗粒度筛选指标,不能替代药化判断。
适用对象仍局限于小分子
论文自己的 limitation 也指出,FlexSBDD 目前只考虑小分子设计,而抗体、肽、核酸等其他药物模态在药物发现中越来越重要;另一个限制是数据规模有限,未来可能需要借助 AlphaFold 3、RoseTTAFold All-Atom 等生成更多 protein-ligand interaction data。
对 AI 制药未来发展的意义
FlexSBDD 的意义不在于已经把生成式药物设计推到可直接工业应用的程度,而在于它清晰指出了一个方向:结构基础生成模型必须从静态口袋生成,走向复合物协同生成。
对 molecular generation 来说,这意味着 ligand prior 不能只学习分子自身分布,还要学习 receptor-induced constraints。一个分子在真空中看似合理,在刚性口袋中看似冲突,但在柔性口袋中可能成立。
对 docking 来说,这提示未来生成与对接可能进一步融合。传统流程是先生成 molecule,再 docking,再评分;FlexSBDD 则把 pose formation 和 receptor adjustment 纳入同一个生成轨迹。未来如果再结合物理能量项、可微 clash penalty、显式水网络和多构象 ensemble,可能会更接近真实结合过程。
对 virtual screening 来说,柔性蛋白生成可以扩大可探索空间,尤其适合 cryptic pocket、induced-fit pocket、耐药突变导致的 pocket remodeling 等场景。但要真正用于筛选,还需要解决计算成本、排序可靠性和实验闭环问题。
对 AI 制药模型评估来说,这篇文章也提供了一个信号:仅报告 Vina Dock 或 QED 已经不够。未来模型需要同时报告几何有效性、相互作用恢复、clash、构象合理性、泛化能力、实验命中率和可合成性。
小结
FlexSBDD 是一篇值得关注的方法论文。它没有停留在让生成分子更像 ligand,而是把问题推进到 protein-ligand complex co-generation:在生成配体的同时,让蛋白口袋做出局部结构响应。它的技术路线比较完整:用紧凑蛋白自由度降低复杂度,用 Riemannian flow matching 统一坐标、旋转、角度和原子类型,用 E(3)-等变标量-向量网络学习三维几何流场,再通过 ApoBind 与 relaxation/repacking 增强 apo-holo 学习信号。
- 真正可靠的 AI 药物生成,最终需要从静态结构拟合走向复合物形成过程建模。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-09,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号