不同差异分析方法拿到的上下调基因影响什么了?


文献里面提到了一个表达量矩阵数据集的两个可能的误差来源,首先是该队列样品收集就耗费了四年所以有批次效应,其次队列里面的性别失衡,这两个问题都是需要解决的。所以研究者们采用了ANOVA model 很严谨的去判别差异基因,方法学如下所示:

采用了ANOVA model
这是一个表达量芯片数据集:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117261,是很经典的的两分组:58 PAH and 25 control lung tissues,然后我也默认走了标准差异分析,以及读取了作者的文献附件里面的差异分析结果,简单的对比了一下:
load('GSE117261/anno_DEG.Rdata')
table(DEG$g)
head(DEG)
DEG=DEG[!duplicated(DEG$name),]
rownames(DEG)=DEG$name
cg = DEG$name[DEG$g!='stable']
my_deg = DEG
load('GSE117261/step1-output.Rdata')
paper_deg =data.table::fread('paper.txt',data.table = F)
paper_deg=na.omit(paper_deg)
head(paper_deg)
str(head(paper_deg))
paper_deg=paper_deg[nchar(paper_deg[,1])>1,]
paper_deg=paper_deg[!duplicated(paper_deg[,1]),]
rownames(paper_deg)=paper_deg[,1]
colnames(paper_deg)=c('Symbol','FC','FDR')
ids= intersect(rownames(my_deg),rownames(paper_deg))
plot(my_deg[ids,'logFC'],paper_deg[ids,'FC'])
plot(my_deg[ids,'logFC'],log2(paper_deg[ids,'FC']))
我和作者的logFC是基本上没有差异的,不过作者在文章附件给出来的是没有log的FC,然后我看了看我们不同方法判别差异分析的统计学显著的上下调基因的一致性,如下所示:
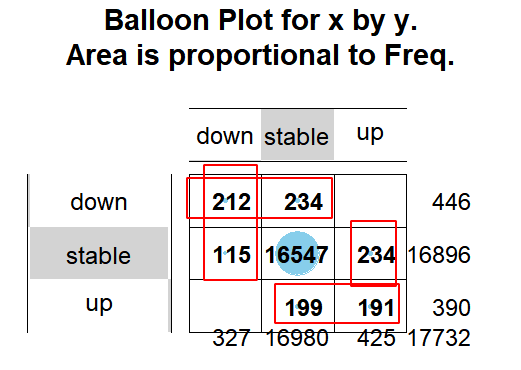
的上下调基因的一致性
在作者的标准里面只需要 false discovery rate [FDR] q value , 0.001 就是统计学显著的差异基因,代码如下所示:
pvalue_t=0.001
paper_deg$g=ifelse(paper_deg$FDR > pvalue_t,'stable', #if 判断:如果这一基因的P.Value>0.01,则为stable基因
ifelse( paper_deg$FC > 1,'up', #接上句else 否则:接下来开始判断那些P.Value<0.01的基因,再if 判断:如果logFC >1.5,则为up(上调)基因
ifelse( paper_deg$FC < 1,'down','stable') )#接上句else 否则:接下来开始判断那些logFC <1.5 的基因,再if 判断:如果logFC <1.5,则为down(下调)基因,否则为stable基因
)
table(paper_deg$g)
而我们的表达量芯片默认的差异分析需要同时卡logFC,所以有火山图如下所示:
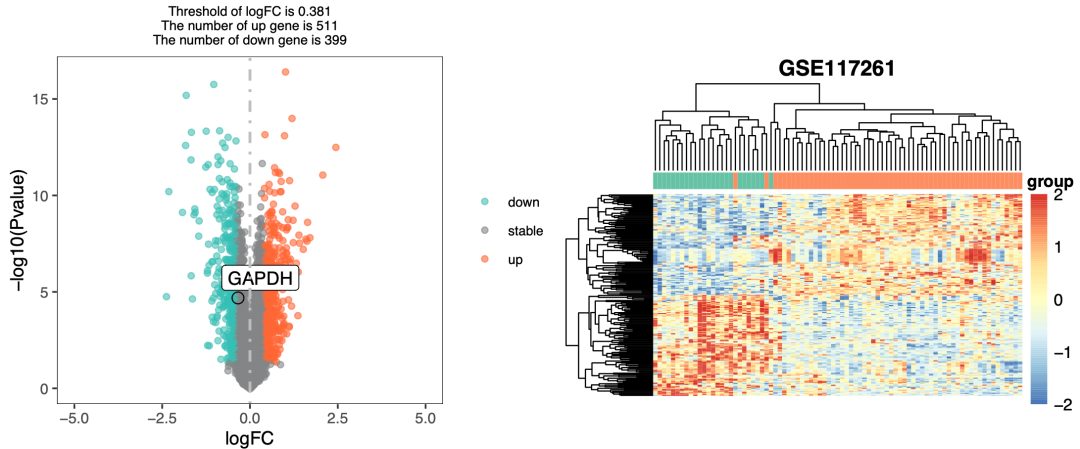
火山图
从火山图可以看到我给出的阈值是很奇怪,因为我采用了 cut_logFC <- with(df,mean(abs(df
logFC)) ) 这个统计学 概念,我们可以细致的检查一下我们的差异分析冲突的基因情况。
df=data.frame(
gene=ids,group=paste0(my_deg[ids,'g'], paper_deg[ids,'g'] )
)
dfl = split(df$gene,df$group)
symbols_list = dfl[-4]
df= melt(dat[unlist(lapply(symbols_list, head)),])
df$group = rep(group_list,each=36)
head(df)
p <- ggboxplot(df, x = "group", y = "value",
color = "group", palette = "jco",
add = "jitter")+facet_wrap(~Var1)
# Add p-value
p + stat_compare_means()
theme_bw()
比如我们先看看3种下调基因:

3种下调基因
最上面的基因是我们两个方法都认为是下调基因,但是中间的是我们的方法判定为下调,但是作者的ANOVA model 反对的,最下面的基因就是反过来的。
然后看看上调基因的冲突情况:
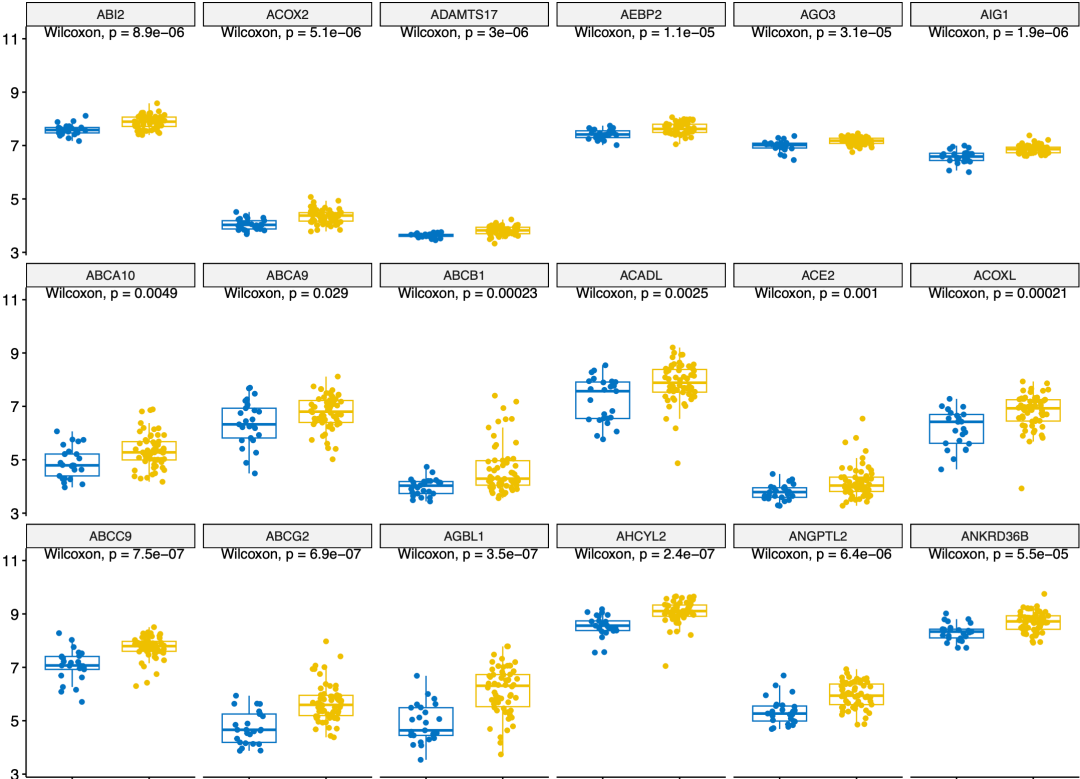
上调基因的冲突
基本上就是一样的情况啦,最上面的基因是我们两个方法都认为是上调基因,但是中间的是我们的方法判定为上调,但是作者的ANOVA model 反对的,最下面的基因就是反过来的。
这个时候,虽然我们搞清楚了,确实是两个方法有冲突,而且冲突的原因就在于对统计学p值的容忍度以及对变化倍数的阈值选择。我们的方法倾向于选择那些变化倍数比较大但是可能统计学不那么显著的,而作者的ANOVA model 恰好就反过来了。
这样的话,我们就产生了6种不同的基因列表,是可以进行生物学功能注释的,代码如下所示:
library(clusterProfiler)
library(org.Hs.eg.db)
library(ReactomePA)
library(ggplot2)
library(stringr)
# 首先全部的symbol 需要转为 entrezID
gcSample = lapply(symbols_list, function(y){
y=as.character(na.omit(AnnotationDbi::select(org.Hs.eg.db,
keys = y,
columns = 'ENTREZID',
keytype = 'SYMBOL')[,2])
)
y
})
gcSample
pro='test'
# 第1个注释是 KEGG
xx <- compareCluster(gcSample, fun="enrichKEGG",
organism="hsa", pvalueCutoff=0.3)
dotplot(xx) + theme(axis.text.x=element_text(angle=45,hjust = 1)) +
scale_y_discrete(labels=function(x) str_wrap(x, width=50))
ggsave(paste0(pro,'_kegg.pdf'),width = 10,height = 8)
可以看到的是作者的方法能发现一些我们的方法被忽视掉的下调的生物学功能,比如胰岛素信号转导通路:
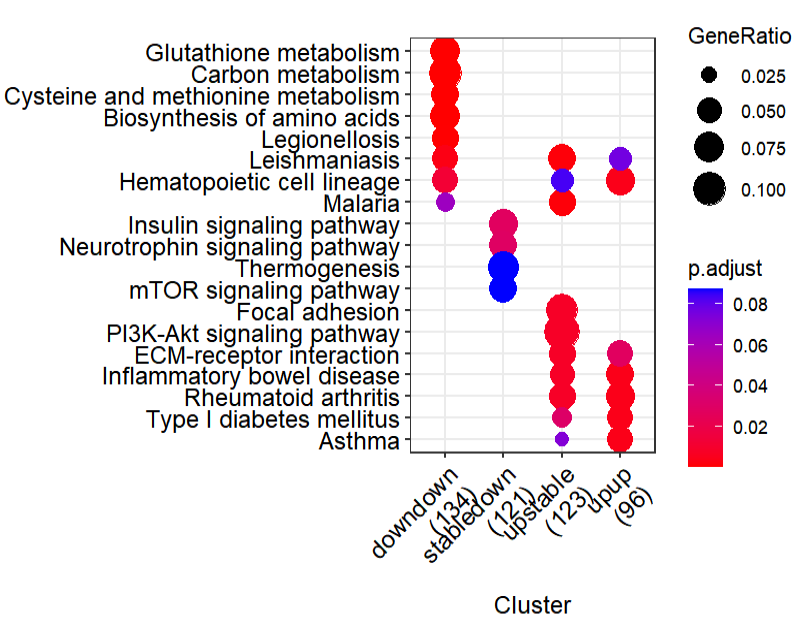
胰岛素信号转导通路
可惜的是这个文章完全就没有使用下调的基因列表或者生物学功能,感兴趣的可以去读一下:Systems Analysis of the Human Pulmonary Arterial Hypertension Lung Transcriptome. Am J Respir Cell Mol Biol 2019 Jun;60(6):637-649. PMID: 30562042
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-04-15,如有侵权请联系 cloudcommunity@tencent.com 删除
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号