做COX生存分析是否需要把连续值变成高低二分组?

做COX生存分析是否需要把连续值变成高低二分组?

生信技能树收到一个粉丝的问题:他做了批量的cox生存分析,发现有一些基因是风险因子,有一些是保护因子。然后进行了一些过滤,并挑一些基因单独拿出来做可视化,但发现有一些基因前面明明pvalue<0.05,但是这里却不具备统计学意义了。经过debug,发现他在批量的时候使用的是基因的连续值,单独可视化的时候用的基因二分组。
那么cox生存分析时,将因素的连续值变成二分组有什么影响呢?
首先我们来问一下人工大模型kimi
Q1:做cox分析的时候是否需要把连续值变成高低二分组?
在进行Cox回归分析时,是否需要将连续变量转化为分类变量(如高低二分组)取决于研究目的和数据特性。Cox回归模型可以处理连续变量,但有时将连续变量转化为分类变量可以提供更明确的临床意义和更易解释的结果。
- 保持连续变量:如果连续变量与生存时间的关系是线性的,或者你希望捕捉变量的细微变化对生存概率的影响,那么保持变量为连续形式可能更合适。
- 转化为分类变量:如果连续变量的某个阈值在临床上具有特别的意义,或者你希望简化模型,那么将连续变量转化为分类变量可能是有益的。例如,基因表达量可以基于中位数分为高表达和低表达两组,然后进行Cox回归分析。
- 寻找最佳截断值:在确定截断值时,可以参考相关文献和指南。如果没有文献支持,可以使用统计方法,如survminer包的surv_cutpoint函数,通过Maximally Selected Test Statistics选择Log-rank检验统计量最大的截断值。
- 模型比较:在某些情况下,比较连续变量和分类变量模型的结果可以提供额外的见解。例如,连续变量的Cox模型可能会得出与分类变量模型不同的风险比(HR)和P值。
总之,选择哪种方法应基于数据的特性、研究目的以及对结果解释的需求。在某些情况下,将连续变量转化为分类变量可以提供更清晰的临床信息,而在其他情况下,保持变量为连续形式可能更合适。
这里人工智能告诉我们,如果变量与生存时间的关系是线性的,就直接使用连续值。如果这个变量的值有很明显的高低值对应的表型差异,可以转为分类变量。
Q1:cox针对基因表达量连续值做生存分析,连续值变成二分组后,结果的显性发生了改变,可能得原因是?
当在Cox回归分析中将基因表达量连续值变成二分组后,结果的显性发生改变,可能的原因包括:
- 线性关系假设:Cox回归要求自变量与因变量之间的关系在转换后应近似线性。如果基因表达量与生存时间的关系是非线性的,将其转化为二分组可能会丢失重要的信息,导致结果的显性发生改变。
- 等比例风险假设:Cox回归的另一个关键假设是等比例风险,即不同组别之间的风险比在时间上保持恒定。如果基因表达量与生存时间的关系随时间变化,那么二分组可能无法准确反映这种变化,从而影响结果的显性。
- 信息损失:将连续变量转化为二分组会丢失变量的精细度,可能导致信息损失。这种信息损失可能会影响模型的预测能力和结果的显性。
- 截断值选择:在将连续变量转化为二分组时,截断值的选择至关重要。如果截断值选择不当,可能会导致分组不合理,从而影响结果的显性。
- 样本量:样本量的大小也会影响结果的显性。如果样本量较小,即使是连续变量,其结果的显性也可能不稳定。在转化为二分组后,如果组内样本量过小,可能会导致统计功效不足,影响结果的显性。
- 模型拟合度:连续变量和二分组变量在模型中的拟合度可能不同。连续变量可能更好地捕捉到基因表达量与生存时间之间的细微关系,而二分组可能过于简化这种关系,导致模型拟合度下降,从而影响结果的显性。
- 统计方法:不同的统计方法和模型设定也可能导致结果的差异。例如,如果连续变量的Cox模型中包含了非线性项或者交互项,而二分组模型中没有,那么结果的显性可能会有所不同。
综上所述,将连续变量转化为二分组后,结果的显性发生改变可能是由于多种因素共同作用的结果。在实际应用中,需要根据具体情况综合考虑,选择合适的统计方法和模型设定。
现在我们直接使用一个案例查看基因的连续值与二分组的Cox回归分析的差异
例如我们使用来自TCGA数据库的BRCA乳腺癌的数据:
rm(list=ls())
library(data.table)
dat <- data.table::fread('inputs/TCGA-BRCA.htseq_counts.tsv.gz',data.table = F)
head(dat[,1:4])
tail(dat[,1:4])
dat = dat[1:(nrow(dat)-5),]
rownames(dat) = dat$Ensembl_ID
a = dat
a = a[,-1]
## 逆转 log
a = as.matrix(2^a - 1)
# 用apply转换为整数矩阵
head(a[,1:4])
tail(a[,1:4])
colSums(a)/1e6
# 普通转录组定量 20m
exp = apply(a, 2, as.integer)
rownames(exp) = rownames(dat)
exp= log(edgeR::cpm(exp)+1)
library(stringr)
head(rownames(exp))
library(AnnoProbe)
library(tinyarray)
rownames(exp) = substr(rownames(exp), 1, 15)
re = annoGene(rownames(exp),ID_type = "ENSEMBL");head(re)
exp = trans_array(exp,ids = re,from = "ENSEMBL",to = "SYMBOL")
head(exp[,1:4])
tail(exp[,1:4])
proj='tcga-brca'
save(exp,file = paste0(proj,".htseq_counts.rdata") )
拿到生存信息:
rm(list=ls())
proj='tcga-brca'
load(file = paste0(proj,".htseq_counts.rdata") )
Group = ifelse(as.numeric(str_sub(colnames(exp),14,15)) < 10,'tumor','normal')
table(Group)
Group = factor(Group,levels = c("normal","tumor"))
print(table(Group))
# 生存分析只需要tumor样品即可
exprSet = exp[,Group=='tumor']
surv = read.delim('inputs/TCGA-BRCA.survival.tsv',header = T)
head(surv)
meta=surv
#去掉生存信息不全或者生存时间小于30天的样本,样本纳排标准不唯一,且差别很大.
k1 = meta$OS.time >= 30
k2 = !(is.na(meta$OS.time)|is.na(meta$OS))
meta = meta[k1&k2,]
meta = meta[,c(
'sample',
'OS',
'OS.time'
)]
colnames(meta)=c('ID','event','time')
meta$time = meta$time/30
rownames(meta) <- meta$ID
s = intersect(rownames(meta),colnames(exprSet))
exprSet = exprSet[,s]
meta = meta[s,]
identical(rownames(meta),colnames(exprSet))
dim(exprSet)
exprSet[1:4,1:4]
kp=apply(exprSet, 1,sd) > 0.1;table(kp)
exprSet=exprSet[kp,]
save(exprSet,meta,file = paste0(proj,".for_survival.rdata") )
## meta的行名和exprSet的列名都是样本名,可以对应起来
# 取第一个基因测试看看
proj='tcga-brca'
load(file = paste0(proj,".for_survival.rdata") )
使用基因的连续值做Cox回归分析
对两万多个基因批量做cox回归分析:
rm(list=ls())
library(survival)
library(survminer)
library(ggstatsplot)
proj='tcga-brca'
load(file = paste0(proj,".for_survival.rdata") )
expression_matrix=as.data.frame(t(exprSet))
expression_matrix$patient_id = rownames(expression_matrix)
clinical_data = meta[,-1]
clinical_data$patient_id = rownames(clinical_data)
colnames(clinical_data) = c('status','time', 'patient_id')
# 数据整合
# 加载必要的库
library(survival)
library(dplyr)
data_integrated <- inner_join(clinical_data,
expression_matrix,
by = "patient_id")
# 单因素Cox回归
univariate_cox <- function(gene_expression) {
formula <- Surv(time, status) ~ gene_expression
cox_model <- coxph(formula, data = data_integrated)
summary(cox_model)
}
# 批量应用单因素Cox回归
results <- lapply(data_integrated %>%
select(-patient_id, -time, -status),
univariate_cox)
results[[1]]
save(results,file = 'results_of_cox.Rdata')
使用基因中位数对样本进行分组做Cox分析
同样也是对所有的基因:
rm(list=ls())
library(survival)
library(survminer)
library(ggstatsplot)
proj='tcga-brca'
load(file = paste0(proj,".for_survival.rdata") )
# 1. prepare data for coxph----
## 批量生存分析使用coxph回归方法
exprSet[1:4,1:4]
phe=meta
head(phe)
mySurv <- with(phe, Surv(time, event))
survival_dat=phe
cox_results <-apply(exprSet , 1 , function(gene){
# gene= as.numeric(exprSet[1,])
group=ifelse(gene>median(gene),'high','low')
group=factor( group,levels = c('low','high'))
if( length(table(group))<2)
return(NULL)
survival_dat <- data.frame(group=group,stringsAsFactors = F)
m=coxph(mySurv ~ group,
# mySurv ~ stage+ group, # 如果有交叉变量
data = survival_dat)
beta <- coef(m)
se <- sqrt(diag(vcov(m)))
HR <- exp(beta)
HRse <- HR * se
#summary(m)
tmp <- round(cbind(coef = beta, se = se, z = beta/se,
p = 1 - pchisq((beta/se)^2, 1),
HR = HR, HRse = HRse,
HRz = (HR - 1) / HRse, HRp = 1 - pchisq(((HR - 1)/HRse)^2, 1),
HRCILL = exp(beta - qnorm(.975, 0, 1) * se),
HRCIUL = exp(beta + qnorm(.975, 0, 1) * se)), 3)
return(tmp['grouphigh',])
})
# 2. specify the value----
cox_results=t(cox_results)
head(cox_results)
cox_results=cox_results[order(cox_results[,'HR'],decreasing = T),]
table(cox_results[,4]<0.01)
table(cox_results[,4]<0.05)
cox_results['DMD',]
save(cox_results, file = 'batch_cox_results.Rdata')
比较两个分析的结果
使用散点图来比较两次分析的每个基因的HR值。
1、首先提取 按照中位数分组后的结果
rm(list=ls())
library(survival)
library(survminer)
library(ggstatsplot)
# cox_results: 按照中位数分组后的结果
load(file = 'batch_cox_results.Rdata')
table(cox_results[,4]<0.01)
table(cox_results[,4]<0.05)
head(cox_results)
cg = strsplit('TAGLN2, IGF2R, LAMP2, TBL1X, ASAP1, DENND5A, SNRK, BCL3 , CEBPD',',')[[1]]
cg = trimws(cg)
kp=cg %in% rownames(cox_results);table(kp)
cg=cg[kp]
cox_results[cg,]
# 提取pvalue和HR值
cox_results_1=cox_results[,c(5,4)]
2、提取 按照连续值分析的结果
# results: 连续值结果
load(file = 'results_of_cox.Rdata')
results[[1]]
# 提取 results: 连续值结果
cox_results_2 = do.call(rbind,
lapply(names(results), function(i){
#i=names(results)[1]
m=results[[i]];m
# 假设cox_model是您的Cox回归模型对象
coef_summary <- m$coefficients
# 提取HR和P值
hr_value <- coef_summary[1, "exp(coef)"] # 假设gene_expression是第一个变量
p_value <- coef_summary[1, "Pr(>|z|)"]
# 打印HR和P值
# print(paste("HR:", hr_value))
# print(paste("P-value:", p_value))
return(c(i,hr_value,p_value))
}))
rownames(cox_results_2)=cox_results_2[,1]
head(cox_results_2)
cox_results_2=as.data.frame(cox_results_2[,-1])
colnames(cox_results_2)=c('hr_value','p_value')
cox_results_2$hr_value = as.numeric(cox_results_2$hr_value)
cox_results_2$p_value = as.numeric(cox_results_2$p_value)
cox_results_2[cg,]
3、两个结果的共同交集
合并两次的分析结果:cox_results_1为分组结果HR值,cox_results_2为连续值结果HR值
# 两个结果取共同交集
ids=intersect(rownames(cox_results_1),
rownames(cox_results_2))
colnames(cox_results_1)
colnames(cox_results_2)
df= data.frame(
cox_results_1 = cox_results_1[ids,'HR'],
cox_results_2 = cox_results_2[ids,'hr_value']
)
df$delt = df$cox_results_1 - df$cox_results_2
head(df)
#
# cox_results_1 cox_results_2 delt
# OR4B1 3.371 0.6709399 2.700060
# LCE1A 3.102 1.2617574 1.840243
# RNU1-3 3.006 1.0652438 1.940756
# RNU1-77P 2.300 1.0091653 1.290835
# RNU1-21P 2.225 0.0988616 2.126138
# MAGEA9 2.148 0.9846482 1.163352
4、绘图比较差异
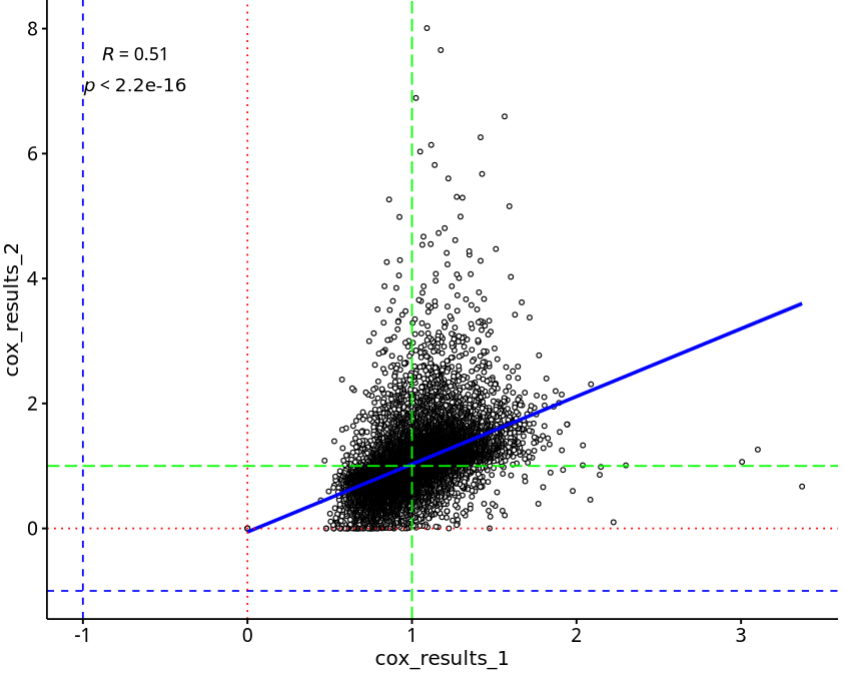
绘制两次分析的HR三点相关性图:
library(ggpubr)
p=ggscatter(df, x = "cox_results_1", y = "cox_results_2",
color = "black", shape = 21, size = 1, # Points color, shape and size
add = "reg.line", # Add regressin line
add.params = list(color = "blue", fill = "lightgray"), # Customize reg. line
conf.int = TRUE, # Add confidence interval
cor.coef = TRUE, # Add correlation coefficient. see ?stat_cor
cor.coeff.args = list(method = "pearson", label.sep = "\n")
)
p=p+geom_hline(yintercept = -1, linetype = "dashed", color = "blue") +
geom_hline(yintercept = 0, linetype = "dotted", color = "red") +
geom_hline(yintercept = 1, linetype = "longdash", color = "green") +
geom_vline(xintercept = -1, linetype = "dashed", color = "blue") +
geom_vline(xintercept = 0, linetype = "dotted", color = "red") +
geom_vline(xintercept = 1, linetype = "longdash", color = "green")
p
结果如下:可以看到两个值的分析结果差异还是挺大的!
腾讯云开发者
