肿瘤病人和正常人有差异表达但并不是说他们的血液层面就很显著


如果是对肿瘤病人和正常人分别取样,取手术后的肿瘤组织,然后去跟癌旁组织或者正常人的对应的部位的组织样品,是很容易看到全局表达量差异的,这一点在tcga的全部癌症都被验证过。而且差异都是合理的,主要是癌症的hallmark在癌症样品里面表达量上调,然后生物学背景告诉我们大家熟知的管家基因理论上并不会差异,详见:housekeeper基因在肿瘤与正常样本中会发生显著差异表达吗?,而且专门有一个专辑:泛癌分析。
但是如果取样的时候,对肿瘤病人和正常人分别取他们的外周血,这个时候就很难看到全局的表达量差异了。这个时候无论是表达量检测手段是什么,表达量芯片,转录组测序,蛋白质组学或者代谢组学,哪怕是单细胞转录组,都有人做过,都很难发现什么全局的差异。但是,大家仍然是会强行找差异然后各种机器学习勉强得到一些结果争取发表出来。比如2025年1月的文献:《LcProt: Proteomics-based identification of plasma biomarkers for lung cancer multievent, a multicentre study》,有多个队列:
- 广州医科大学第一附属医院(1STGMU)队列(GMU队列的一个亚队列)的127名参与者的数据,其中包括106名肺癌患者和21名良性肺部疾病患者
- 即国家呼吸医学中心 (NCRM) 队列,其中包括 75 名肺癌患者和 26 名良性肺部疾病患者。
- 天津市临床多组学重点实验室(TKLCM),30名肺癌患者和16名良性肺部疾病患者进行外部验证。
首先呢,有肿瘤病人和非肿瘤这个疾病的人,可以差异分析,任务#1。然后呢,肿瘤病人里面有lymph node metastasis的临床分组信息,是任务#2 。最后,肿瘤病人有tumour‒node‒metastasis (TNM) staging临床信息,也是可以差异分析,是任务#3。
差异分析之前,其实需要看表达量矩阵的3张图。很早以前我就在生信技能树的教程:《你确定你的差异基因找对了吗?》提到过,必须要对你的转录水平的全局表达矩阵做好质量控制,最好是看到标准3张图:
- 左边的热图,说明我们实验的两个分组,normal和npc的很多基因表达量是有明显差异的
- 中间的PCA图,说明我们的normal和npc两个分组非常明显的差异
- 右边的层次聚类也是如此,说明我们的normal和npc两个分组非常明显的差异
如果分组在3张图里面体现不出来,实际上后续差异分析是有风险的。这个时候需要根据你自己不合格的3张图,仔细探索哪些样本是离群点,自行查询中间过程可能的问题所在,或者检查是否有其它混杂因素,都是会影响我们的差异分析结果的生物学解释。
如果看这个文献里面的3个表达量矩阵的样品异质性图(pca,tSNE或者Umap),可以很明显的看到居然是恶性的癌症病人样品跟良性疾病的样品差异很难区分,就因为取样是Peripheral blood samples,反而是TNM staging临床信息分组很明显:
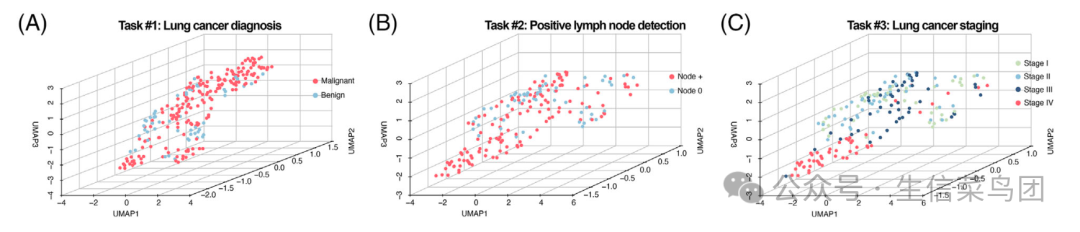
样品异质性图
直接使用使用“limma”包找差异表达蛋白 (DEP)即可,使用统一的阈值:绝对对数倍数变化大于 1 且调整后的p值小于 0.05 的蛋白质视为差异表达。
- 任务#1 的 300 个被确定为 DEP,包括 255 个显着上调的蛋白质和 45 个显着下调的蛋白质
- 任务#2 的 920 个被确定为 DEP,包括 716 个显着上调的蛋白质和 204 个显着下调的蛋白质
- 任务#3,总共 2251 个蛋白质被识别为 DEP。(根据stage的1,2,3,4进行两两组合的多次差异分析)
这些差异分析的基因数量,基本上就可以在前面的样品异质性图(pca,tSNE或者Umap)看出来的,如下所示每次差异分析都可以显示出来统计学显著的上下调基因:
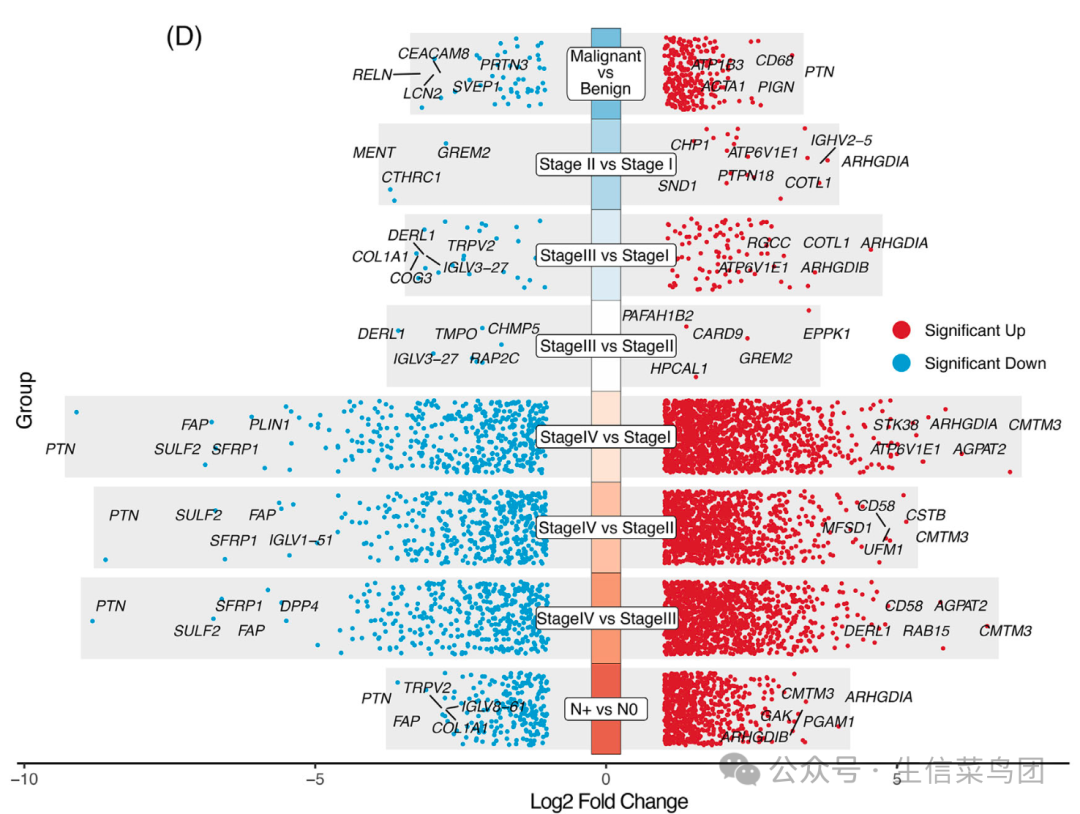
可以看到,其实主要的差异都是来源于stage4跟其它的stage的比较,在前面的样品异质性图(pca,tSNE或者Umap)也可以看到是stage4跟其它的stage出现了泾渭分明的分隔。
有意思的是,研究者们其实并不是仅仅是取样了blood,在文献里面写了是:Peripheral blood samples before and after treatment, tumour tissue, pair para-cancerous tissue and biopsy tissue samples were planned to collect based on the treatment protocols.
但是为什么不做真正的癌症组织样品和癌旁样品呢?他们的差异首先是更大一点,其次也更直接啊!
因为作者并没有公开他们的蛋白质组学表达量矩阵信息,所以没办法针对作者自己的数据进行图表复现!但是有很多类似的实验设计可以试试看跟这个研究做的对比!
比如:
- 数据集: GSE43580,是 51 AC1 (adenocarcinoma stage I), 36 AC2, 34 SCC1(squamous cell carcinoma,) and 39 SCC2 samples
- 文献PMID: 19951989:取样是 peripheral blood mononuclear cell (PBMC) ,来源于 137 patients with NSCLC tumors and 91 patient controls with nonmalignant lung conditions
- 数据集GSE20189 ,取样是 lung tissue and peripheral whole blood (PWB) from adenocarcinoma cases and controls,是153 subjects (73 adenocarcinoma cases, 80 controls)
- tcga的luad和lusc数据集,We used 226 samples in which cancer metastases were annotated and divided them into two categories, namely, lung cancer metastasis and non-metastasis. 来源于文献:https://doi.org/10.1016/j.compbiomed.2022.106490
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-01-18,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号