构建和使用整合单细胞图谱,需要考虑哪些因素?



Basic Information
- 英文标题:Considerations for building and using integrated single-cell atlases
- 中文标题:构建和使用整合单细胞图谱的考虑因素
- 发表日期:13 December 2024
- 文章类型:Review Article
- 所属期刊:Nature Methods
- 文章作者:Karin Hrovatin | Malte D. Luecken
- 文章链接:https://www.nature.com/articles/s41592-024-02532-y
Abstract
Para_01
- 单细胞技术的迅速采用为构建整合多个实验室多样化数据集的单细胞‘图谱’提供了机会。
- 这些图谱可以作为分析和解释当前及未来数据的参考。
- 然而,很明显,不同的图谱构建方法存在差异,而这些差异的影响往往不明确。
- 在这里,我们回顾了当前的图谱文献,并提出了构建和使用图谱时需要考虑的因素。
- 重要的是,我们发现并不存在适用于所有情况的单一图谱构建协议,而是讨论了特定情境下的考虑因素和工作流程,包括图谱的概念化、数据收集、整理与整合、图谱评估以及图谱共享。
- 我们进一步强调了集成图谱在分析新数据集和获取超越单一数据集所能提供的生物学见解方面的优势。
- 我们对当前实践的概述及相关建议将提高未来图谱的质量,推动向统一的、基于参考的单细胞生物学理解的转变。
Main
Para_01
- 了解组织的细胞组成及其在个体间的差异对于理解健康与疾病至关重要。
- 单细胞技术通过使研究人员能够以前所未有的规模和分辨率研究组织,推动了我们对细胞异质性的理解取得重要进展。
- 然而,尽管每个研究中的单细胞数据集数量和测序细胞数量不断增加,目前每项研究中采样的个体中位数仍然不超过14个(图1)。
- 此外,单独的研究存在与特定研究相关的偏差,例如队列特征、样本处理以及单细胞技术的选择。
- 将多项研究整合到一个单一资源中,这里称为‘图谱’,可以使研究人员克服这些特定研究的偏差,并捕获更多个体,更全面地描绘细胞多样性。
Fig. 1: Single-cell dataset size trends over time.
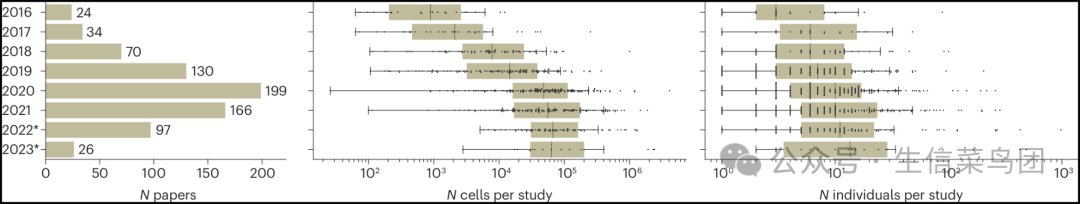
- 图片说明- 左侧为每年发表的包含单细胞数据的论文总数;中间为随时间推移每个数据集中的细胞数量;右侧为每篇单细胞论文中包含的个体数量(补充方法)。- 出版物列表来源于一个精心整理的单细胞研究数据库。- 2021年之后的数据(用星号标记)可能不完整,因此论文数量可能被低估。- 对于两个箱线图,箱子表示中位数和四分位距。- 须线延伸到最远的非异常值数据点。- 单个数据点以点的形式显示。
Para_02
- 许多研究计划,包括人类细胞图谱(HCA)和人类生物分子图谱计划(HuBMAP),旨在创建人体的单细胞图谱。
- 目前已有的单细胞图谱涵盖了来自小鼠、人类或两者皆有的各种组织,几乎完全由转录组学数据组成(有关可用图谱及其特征的概述,请参见补充表1)。
- 这些图谱已被用于解决一系列生物学问题和研究挑战。
- 例如,它们为细胞类型定义和疾病特异性细胞状态提供了共识,揭示了群体在大规模上的异质性,帮助分析新数据集,并为研究设计提供指导。
- 现有的单细胞图谱已经展现出推动我们对健康与疾病中人体组织理解的潜力。
Para_03
- 作为社区资源,整合的单细胞参考图谱应遵循特定标准。
- 首先,图谱旨在作为细胞类型命名法社区讨论的基础,因此应尽可能反映当前细胞类型的定义共识。
- 其次,从图谱中得出的发现需要具有普适性,并代表跨研究的共识,这就要求包含众多且多样化的数据集,涵盖大量个体。
- 因此,基于个别数据集的近期大规模努力在未来将成为构建多数据集图谱的良好基础。
- 普适性还要求记录与数据集或样本相关的偏差,例如数据生成地点和协议,并在可能的情况下将其从图谱中移除。
- 第三,图谱需要包含广泛的细胞注释以及样本和主体元数据,以供未来研究和分析使用。
- 最后,图谱必须可靠,确保从中得出的发现不是基于数据中的伪影或注释中的错误,这需要严格的质量评估。
- 因此,这类图谱可以像参考基因组和其他组学参考一样,作为新数据集比较的‘正常’基准,用于解决分子生物学和医学中的核心问题。
Para_04
- 尽管构建图谱的过程复杂且要求高,但目前尚缺乏对构建图谱步骤及其相关考虑因素的清晰概述。
- 此外,图谱在研究中的潜在应用才刚刚开始被探索。
- 在此,我们回顾了以往参考图谱的构建方式,并提出了指导原则,以确保未来图谱的构建与共享具备高质量和广泛的应用机会。
- 我们还探讨了未来图谱如何扩展和更新,以跟上新发现的步伐。
- 最后,我们提供了图谱使用案例的全面概述。
- 总体而言,我们期望这项工作能够推动以图谱为重点的计划(如HCA、HubMAP等)的发展,从而促进单细胞领域向跨数据集、群体水平的参考图谱迈进。
[div_box]
Building an integrated reference atlas
Para_01
- 构建一个综合图谱需要生物学和计算领域的专业知识,并对图谱进行迭代优化。
- 这一过程可以分为以下几个步骤(图2),以下将详细讨论:准备阶段,包括确定研究重点和选择数据集;数据预处理,包括元数据协调和质量控制;数据整合;图谱评估与重新注释;以及图谱共享与扩展。
Fig. 2: Workflow for building reference atlases.
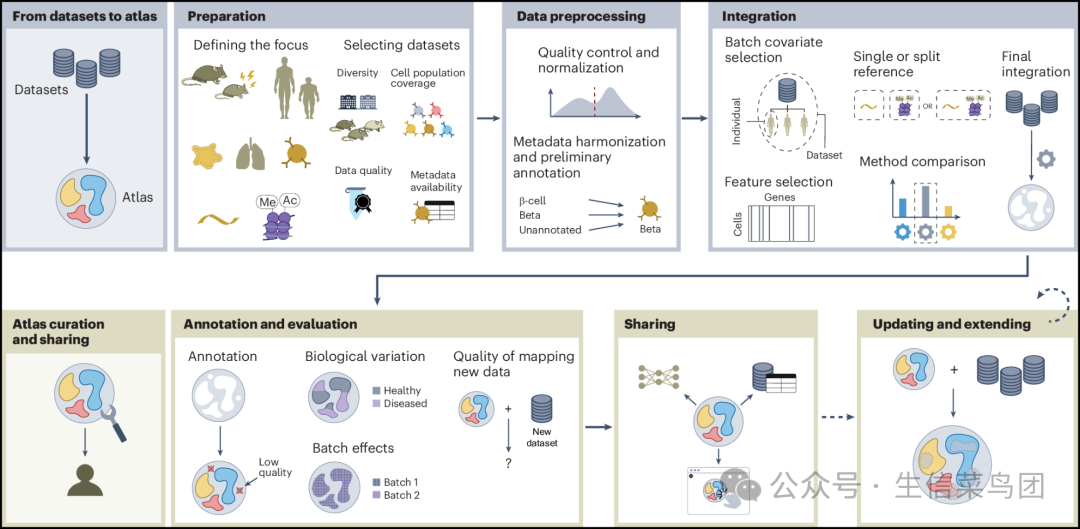
- 图片说明- 图谱构建过程包括从单个数据集构建图谱(上部分;包括数据准备、预处理和整合),以及在图谱构建完成后对其进行管理和共享(下部分;包括图谱注释与评估、共享,以及对图谱中捕获的数据进行更新和扩展)。
Atlas preparation
图集准备
Para_01
- 对图谱的预期下游用途决定了在构建图谱时应做出哪些技术决策,因此在准备阶段就应考虑图谱的目标。
- 例如,如果要构建一个能够模拟年龄对分子表型影响的图谱,理想情况下应包含儿科样本。
- 因此,在开始构建之前,确定图谱的重点至关重要。
- 同样,所选数据集应仔细筛选,以与图谱的重点保持一致并最大化其质量。
- 下面,我们将讨论这两个过程中的重要考虑因素。
Defining the focus
确定关注的重点
Para_01
- 可能希望制作尽可能通用的图谱,整合跨技术、器官或物种的数据。
- 然而,这最终可能会降低其实用性,因为去除强烈的批次效应通常也会导致生物变异的过度损失。
- 相反,必须在一开始选择图谱的重点,以确保最终的图谱最适合预期的下游应用。
- 虽然大多数图谱旨在全面理解单一器官,但细胞类型特异性的图谱可以为影响多个组织的细胞类型特异性疾病提供洞察。
- 此外,尽管一些图谱仅关注健康成年样本,但包含多种条件(如疾病或发育阶段)对于跨条件比较至关重要。
- 同样,包含动物或体外模型系统的图谱对于评估模型的实用性至关重要,并且还可以用于补充稀缺的人类数据。
- 最后,多组学图谱可以通过更广泛的分子特征集提高分辨率和可靠性。
- 我们在补充注释1中提供了关于定义图谱重点的进一步指导。
Selecting datasets
选择数据集
Para_01
- 一旦明确图集的目标,就必须选择要包含的数据集,这将重要地决定图集的质量和实用性。
- 对于某些图集,数据稀缺性需要在数据集选择时放宽标准,而在其他情况下,并非所有符合图集目标的数据集都适合被纳入。
- 在此我们讨论选择数据集的关键考虑因素。
Number and technical diversity of datasets
数据集的数量和技术多样性
Para_01
- 包含大量数据集不仅对于捕捉细胞类型和组织表型的变异性很重要,而且对于覆盖多样化的技术变异性也很重要,例如样本处理协议和测序技术。
- 这可能会扩大所涵盖的细胞类型的范围,因为不同的技术能够更好地捕获不同的细胞类型,同时为集成方法提供更多的训练数据以区分生物效应和批次效应,并在下游分析中评估研究结果的可重复性。
- 大量的数据集还允许移除那些后来发现无法良好整合的数据集,而不会显著减少图谱的规模。
- 然而,增加数据集的数量将导致更长的数据整理和预处理时间,并可能最终导致计算资源需求过高而难以承受。
Metadata availability
元数据可用性
Para_01
- 样本级别的元数据(例如个体的年龄或健康状况)和细胞级别的元数据(例如细胞类型标签)对于图谱构建过程中的许多步骤以及其使用都非常重要。
- 与技术变量相关的元数据,例如组织采样技术(例如,尸检和活检样本),将有助于在图谱构建和评估过程中区分实验偏差与生物学驱动的信号。
- 此外,详细的供体元数据(例如年龄、性别、体重指数、吸烟史或疾病阶段及治疗史)将使图谱更具广泛适用性,特别是对于理解个体间差异非常有帮助。
- 同样,细胞级别元数据的可用性,主要是细胞类型标签,可以在多个方面辅助图谱构建过程,包括质量控制(例如双重细胞的标签)、整合(对于使用细胞类型标签的方法)以及图谱评估和使用。
Demographic diversity
人口多样性
Para_01
- 为了全面表示给定组织,图谱应涵盖人类群体在年龄、性别、遗传祖先及其他人口统计变量方面的多样性。
- 同样的原则也适用于其他类型的生物变异,例如多种小鼠模型或品系以及各种环境条件。
- 增加样本的多样性可以使基于图谱的研究结果更具普遍性,并可能实现对患者群体等进行分层。
Cell-type coverage
细胞类型覆盖范围
Para_01
- 理想情况下,图谱应全面代表感兴趣器官或细胞区室中的所有细胞类型多样性,这可以通过多种方式选择数据集来实现。
- 当器官不同部分的细胞组成差异较大时,应覆盖不同的解剖位置。
- 同样,组织取样方式(例如,通过刷取、活检或手术切除)会影响样本中捕获的细胞类型,解离协议也会产生影响,某些细胞类型只能通过特定协议检测到。
- 空间分析方法不受组织解离协议的影响,可用于确定组织中应检测到的细胞类型。
Study design
研究设计
Para_01
- 在构建图谱时,将与批次相关的变量从感兴趣的生物信号中解开是实现有意义的下游分析的关键。
- 然而,如果没有适当的研究设计,批次校正方法可能会无意中在去除批次效应的同时也移除了生物信号。
- 例如,如果一个数据集仅包含来自患有某种疾病(该疾病在其他任何数据集中都未出现)的供体样本,在整合过程中可能会无意中将这种特定于疾病的生物学特征误认为是特定于数据集的批次效应,并将其从整合结果中移除。
- 因此,理想情况下,每个感兴趣的生物群体应在多个数据集中有所体现,并且每个数据集应包含多种条件,以便能够将数据集特定的批次效应与感兴趣的效应区分开来。
Data quality
数据质量
Para_01
- 将高质量的数据集纳入图谱中将提高下游分析的可靠性,并且还将简化数据集成的过程。
- 数据集在质量上可能存在显著差异,例如,在测序深度、每个细胞的线粒体读数比例以及细胞类型注释的详细程度方面。
- 例如,由于测序深度较低导致细胞分辨率较低的数据集已被证明与其他数据集的整合效果较差
[div_box]
Data harmonization and preprocessing
数据协调与预处理
Para_01
- 对于要联合分析的数据集,每个数据集中提供的主体、样本和细胞的元数据必须在各个数据集之间保持一致编码,并且计数数据应以相似或相同的方式进行计算预处理。
- 单独的数据集通常以不同的方式预处理,并使用不同的元数据命名法,这分别导致了批次效应并阻碍了下游解释。
- 虽然元数据必须始终统一,但目前尚不清楚预处理中的具体差异如何影响最终的图谱。
- 下面我们将讨论在优先考虑不同图谱构建步骤的协调时需要考虑的因素。
Data preprocessing
数据预处理
Para_01
- 将 FASTQ 测序数据预处理为计数矩阵是单细胞测序数据分析预处理的第一步。
- 由于计数矩阵比原始 FASTQ 文件更容易共享和整合,因此通常使用计数矩阵来构建图谱,但生成计数矩阵的方式不一致可能导致数据中出现批次效应。
- 尽管重新对所有数据进行比对可能并不总是可行的,但使用基因标识符而不是基因名称已经可以减轻批次效应的影响。
- 一旦生成了计数矩阵,就需要从数据中去除低质量的液滴。
- 在某些情况下,数据集生成者可能已经完成了这一操作,而去除低质量液滴的具体方法可能会导致不同数据集之间的差异。
- 此外,选择仅标注而不移除低质量液滴的决定可以使质量控制转移到新数据集中,尽管这也可能影响数据集的整合。
- 最后,在跨数据集时,应以相同方式对计数进行归一化和校正,例如针对环境 RNA 进行校正,并考虑归一化方法的适用性和可扩展性。
- 有关预处理的相关考虑详见补充注释 2。
Harmonizing sample and subject metadata
协调样本和主体元数据
Para_01
- 样本和主体元数据对于构建图谱的过程以及对图谱的下游分析都至关重要。
- 然而,由于数据集之间的命名法常常不一致,这使得它们的使用变得具有挑战性。
- 因此,所有来自单个数据集的元数据都应该映射到标准化的类别中。
- 对于许多形式的元数据,标准化的术语已经以本体的形式存在,例如疾病、祖先或单细胞协议的相关本体。
- 由于这些现有的本体是由专家构建的,遵循它们通常会提供比手动设置分类更好的分类和命名系统。
- 此外,这将促进领域内的未来交流和比较。
- 单细胞数据平台,如 CELLxGENE 和 HCA 数据存储库,已经符合这些本体标准。
Para_02
- 对于人类元数据,理想情况下还应跟踪这些数据是基于自我报告、医生分配还是基因组信息。
- 由于关于种族和遗传祖先分类的标准仍在不断发展,因此在将这些数据统一到预定义的、可能更广泛的组之前,以‘原始’形式收集这些数据是有用的。
- 或者,从原始测序数据中推断性别或祖先的数据驱动方法可以补充报告的元数据。
Harmonizing cell-type annotations and annotating unlabeled datasets
协调细胞类型注释并标注未标记的数据集
Para_01
- 初步的、作者提供的细胞类型注释可以在许多方面带来好处。
- 首先,它们可以帮助数据整合本身,因为一些数据整合方法允许使用细胞类型标签来指导整合过程。
- 其次,这些标签有助于在数据整合后评估图谱的质量,但必须确保不使用相同的标签来进行整合和评估,以避免评估偏差。
- 第三,与原始标签的比较可以用来评估共识重新注释在图谱中的影响(详见‘探索图谱内的信息’部分)。
Para_02
- 对于上述任务,拥有一组一致的细胞类型标签至关重要。
- 由于研究在细胞类型命名法和注释分辨率上常常不一致,因此将所有注释映射到一个通用的细胞类型命名参考中是有帮助的。
- 当原始注释具有足够的分辨率时,这可能会生成高质量的初步图谱注释。
- 细胞类型参考可以是‘分层的’,从而适应来自不同数据集的不同分辨率的注释。
- 由于手动协调细胞类型标签工作量大,自动构建这样的细胞类型层次结构可以辅助这一过程,例如使用 CellHint。
- 此外,社区资源如细胞注释平台(CAP)正在开发中,以促进作者指导的、基于共识的细胞注释,并为细胞类型和状态定义标签同义词。
Para_03
- 如果作者提供的细胞类型注释不可用或质量不足,可以专门为图谱对数据进行注释。
- 由于对每个数据集单独进行手动注释非常耗时,因此可以仅对具有代表性的数据子集进行注释。
- 或者,可以使用自动注释结合人工校正来对单个数据集进行注释,然后再进行整合,这也可以与现有的注释相结合。
[div_box]
Data integration
数据集成
Para_01
- 任何图谱构建项目的核心都是数据的整合,这涉及通过多种方法计算去除批次相关的变异。
- 整合使得所有数据能够在共享空间中进行联合分析,这种分析基于生物学信号而非批次特定的转录组学伪影。
- 下面我们将描述图谱级别数据整合的几个重要方面。
Determining the level of integration by setting the batch covariate
通过设置批次协变量确定集成水平
Para_01
- 所有数据整合方法的目标都是根据预先定义的批次协变量识别并随后消除特定批次的转录组偏移。
- 批次协变量的选择将极大地影响从图谱中去除哪些变异以及整合后的图谱会呈现何种面貌。
- 需要注意的是,被确定为‘批次效应’的变异本质上是主观的,反映了图谱构建者认为不需要的内容。
- 因此,批次协变量的选择应与图谱的范围保持一致。
Para_02
- 批次效应可能发生在数据集、受试者甚至样本层面。
- 许多实验和预处理因素主要在数据集层面上变化,例如组织分离协议、单细胞化学或参考基因组。
- 因此,在数据集层面进行批次校正已经可以消除大部分由技术因素引起的变异。
- 即使从数据中去除了所有数据集层面的批次效应,由于人工因素导致的转录组变异的其他来源仍可能存在于样本和受试者层面。
- 例如,样本可能经历了不同的处理过程,从而引起不同的转录组变化;在同一测序通道上进行测序的细胞(这些细胞可能来自单一组织样本或多个组织样本,例如在多重测序时),可能会受到特定的技术影响。
- 此外,个体之间可能存在与受试者相关的批次效应,例如由于不同的死后间隔时间。
- 为了去除这些批次效应来源,需要将样本和/或受试者设置为批次协变量。
- 值得注意的是,某些方法允许同时使用多个批次协变量,从而实现嵌套(数据集-样本)或组合(数据集-检测)批次效应设计。
Para_03
- 技术协变量对批次效应的贡献程度可能有所不同。
- 例如,如果某项研究中的样本由多个研究所生成,这些样本可能会或不会表现出与研究所相关的批次效应。
- 同样,批次协变量可能以不同的强度影响个别细胞类型。
- 通过计算特定技术协变量解释的方差百分比,可以定量估算协变量对批次效应的贡献。
- 或者,可以使用分类器检查哪些样本组能够与其他数据集部分轻松区分,从而确定数据集是否需要以及如何划分为单独的批次。
- 最近的一些努力还试图自动化批次选择过程。
Para_04
- 在批次协变量的选择中,主要的陷阱在于整合过程中可能会去除与所选批次协变量共变的生物学信号。
- 当批次直接对应于感兴趣的生物学变量时,尤其会面临挑战。
- 例如,在整合不同类器官协议、物种、器官位置或患有不同疾病的患者样本的数据集时,就会出现这种情况。
- 在此类情况下,去除数据集层面或样本层面的变异可能会从整合图谱中移除感兴趣的方差(例如,特定于协议的状态),从而阻碍下游分析。
- 在这种情况下,可以选择一个更粗粒度的批次协变量,该协变量不会直接与感兴趣的协变量混淆,例如选择数据集而非样本。
- 然而,需要注意的是,一些现有的图谱即使在使用样本作为批次协变量时,仍然显示出样本层面变异的生物学保留。
Selection of genes for data integration
数据整合的基因选择
Para_01
- 类似于其他单细胞RNA测序(scRNA-seq)降维技术,数据集成可以通过在一部分基因上进行操作而获益。
- 这些好处包括提高信噪比、去除与图谱范围无关的非信息性信号以及提高计算效率,从而改善数据整合效果。
- 然而,在去除基因时,必须牢记现有的集成模型无法适应后期添加特征(基因),例如对将来映射到图谱的样本可能重要的特征。
Para_02
- 目前,基因选择最常见的做法是选择‘高变基因’,即在数据中表现出比根据其平均表达水平预期更高的方差的基因。
- 大多数图谱会选择 2,000 到 5,000 个基因,对于涵盖范围更广的图谱,则倾向于选择更多数量的基因。
- 通常,基因被选为在各个单独批次内变异的基因的交集,以避免选择因批次效应而变化的基因。
- 此外,还提出了几种旨在提高基因选择稳健性或生物学意义的方法。
- 这些方法包括去除受批次影响或与质量指标相关的基因,并选择与感兴趣信号相关的基因,例如单个细胞谱系、稀有细胞类型或疾病(补充注释 3)。
- 鉴于所提出方法的多样性,优化图谱构建的这一步骤很可能具有很大的潜力。
Selecting an optimal data integration strategy
选择最优的数据集成策略
Para_01
- 集成方法及其参数的选择将对集成结果产生重大影响。
- 由于不同的集成方法在不同场景下表现最佳,因此为当前数据选择最优方法和参数设置非常重要,例如使用现有的集成基准测试平台。
- 方法 scANVI、Scanorama 和 scVI 已被证明在复杂的集成任务中表现良好,因此如果时间限制不允许进行广泛的方法基准测试,可以优先考虑这些方法。
- 此外,关于多模态数据的集成策略在补充注释 4 中进行了讨论。
Para_02
- 整合过程还涉及决定将哪些可用数据包含在单一的综合表示中。
- 在某些情况下,批次效应过于强烈,无法在保留所需生物信息水平的同时被去除。
- 在这种情况下,图谱可能会被拆分为多个部分,例如,为每个物种、每个谱系创建一个子图谱,或者针对细胞与细胞核分别创建图谱。
- 值得注意的是,最近的研究努力致力于促进更多生物学和技术多样性数据集的整合。
- 尚不清楚全局整合策略与拆分方法相比如何。
- 尽管由多个集成表示组成的图谱可能提供更好的分辨率,但它们的用户友好性较低,需要对每个子图谱进行单独分析和比较。
Para_03
- 一些图谱采用将数据分为核心数据集和扩展数据集的方法,核心数据集通常是整合的,而扩展数据集则通过查询到参考的映射方法映射到核心数据集上。
- 这些方法使得在去除批次效应的同时,能够将新数据投影到现有的参考数据集上成为可能。
- 在某些情况下,基于生物学特性分离核心数据集和扩展数据集可能是有利的,例如,仅使用健康成人的数据作为参考,以在整合过程中最小化生物混淆因素的影响,从而更轻松地学习批次效应。
- 核心-扩展方法还提供了更大的灵活性来适应和扩展一个图谱。
- 给定一个固定的核心参考,数据集可以独立映射到核心上。
- 然而,由于图谱模型的核心从未用新映射的数据进行更新,这些数据集中新的生物学变异可能无法被充分捕捉,或者模型可能无法充分去除新的批次效应。
- 此外,大多数目前可用的查询到参考的映射方法仅与特定的整合方法或模型兼容(例如,Symphony 与 Harmony,scArches 与基于条件自动编码器的方法)。
- 在选择数据整合方法时,应考虑这些兼容性,以便未来的用户能够更容易地将新数据映射到图谱中(‘将新的单细胞数据投影到图谱空间’)。
Para_04
- 集成方法的性能可能差异很大,因此应为给定的数据集集合选择最佳的集成方法。
- 重要的是,通过可视化检查集成结果(例如,使用均匀流形近似和投影(UMAP))来评估性能可能会产生误导,并且难以应用于大量的集成输出。
- 因此,应将视觉检查与定量指标结合使用以评估集成质量。
- 一些指标旨在量化批次效应是否被有效去除,或在数据集成过程中生物变异是否得以保留。
- 由于不同的指标衡量集成表示的不同方面和分辨率,并具有其特定的优点和局限性,因此选择使用哪些指标非常重要。
- 例如,既应包括依赖先验生物学知识的指标(例如,细胞标签),也应包括与其无关的指标。
- 值得注意的是,对单个图谱的数据集成方法进行基准测试是非常耗费资源的。
- 因此,对于包含大量细胞的图谱,可以使用数据的一个子集来进行集成方法的基准测试。
- 我们在补充注释5(框4)中提供了有关集成基准指标和数据子集化的更多详细信息。
[div_box]
Atlas evaluation and reannotation
图集评估与重新标注
Para_01
- 图谱的质量对于其作为参考的实用性至关重要。
- 由于如上所述的自动化整合方法评估无法保证表现最优的方法足以满足图谱使用的质量要求,因此应辅以手动图谱评估。
- 这还包括图谱级别的细胞类型重新注释,它不仅用于评估整合表示的质量,还为下游分析提供基础。
Evaluation of overall atlas representation quality
整体图集表示质量的评估
Para_01
- 最终的图谱评估必须基于先前的生物学知识进行。这确保了图谱能够准确反映数据中的生物学信息,并且批次效应已得到充分消除,如下文所述。
- 评估步骤是优化图谱表示的最后一道检查点(图3a-d),并可能导致重新审视和调整早期的图谱构建步骤(图3e-k)。
Fig. 3: Workflow for evaluating and improving the atlas.
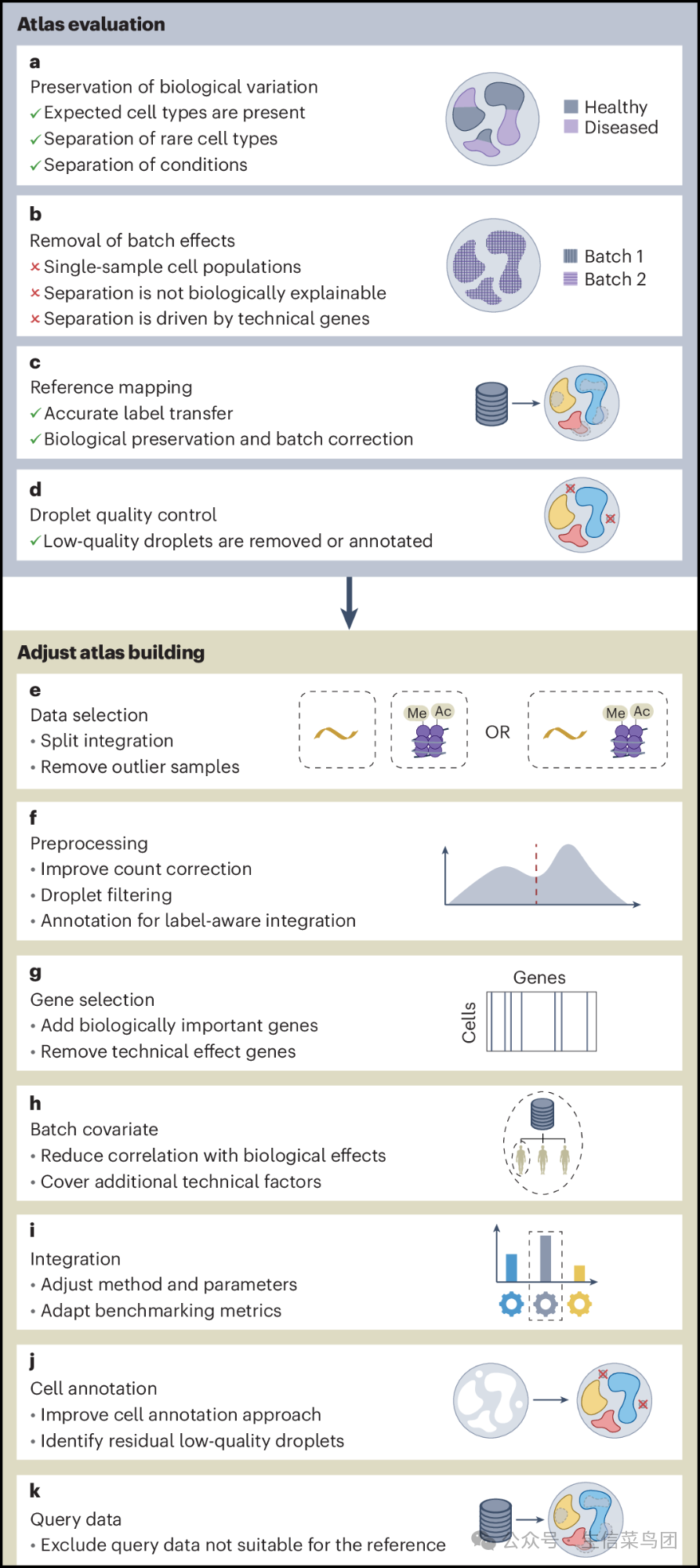
- 图片说明- 在进行下游任务之前,必须从不同角度评估整合图谱的质量。- 该评估应涵盖生物信息的保留(a)、批次效应校正(b)、参考映射(c)以及图谱的细胞组成(d)。- 如有必要,可以通过修改图谱构建工作流中的各个步骤(e-k)来提高图谱质量(图2)。
Para_02
- 为了从图谱中获得新的见解,必须确保数据整合过程中没有移除关键的生物学信息(图3a)。
- 这可以通过评估已知生物学因素(如细胞类型、年龄或疾病)在整合表示中细胞的共现或分离情况来实现。
- 第一步是根据表示中存在的与已知细胞类型相对应的聚类,评估其预期的生物学效应。
- 例如,稀有和过渡期细胞聚类由于过度整合,通常与其他细胞群体合并。
- 上述描述的整合基准指标可以进一步用于突出显示整合质量较差的细胞子集,从而需要进一步的手动探索。
Para_03
- 在分析细胞类型聚类中由生物学驱动的细胞状态和亚型时,必须谨慎,不要将由批次效应引起的分离解释为生物学差异。
- 因此,基于特定协变量(例如疾病)的细胞表示的分离应在重复样本和数据集中得到支持,并且细胞群体也应通过特定标记的表达来区分。
Para_04
- 为了确保下游分析由生物学因素而非批次效应的残余变化驱动,有必要评估批次效应是否已从图谱中成功去除(图3b)。
- 为了全面彻底地评估图谱中剩余的批次效应,需要检查整合表示是否存在由技术效应驱动的细胞分离现象。
- 这些现象包括无法用生物学解释的样本特异性或数据集特异性聚类。
- 一种识别由批次效应驱动的细胞分离的方法是,通过已知技术效应基因的表达与聚类分配的相关性进行分析,这些基因通常具有样本特异性。
- 这包括环境基因、与组织处理相关的基因(如因解离和长时间处理而诱导的压力基因),或者在整合单细胞和单核数据时,已知在两种检测方法之间差异表达的基因(如线粒体基因)。
- 然而,需要注意的是,这些基因也可能参与生物学相关的进程,例如疾病相关的细胞变化。
Para_05
- 尽管图谱中一小部分样本或主体,或者单一数据集可能由于该数据子集中的批次效应较强而未被很好地整合,但图谱整体质量仍可能是优秀的。
- 视觉检查有时已经能够突出显示图谱中整合不佳的子集。
- 此外,应使用评估细胞群体内批次混合情况的指标来识别异常值。
- 确定整合效果下降的原因非常重要,因为它可能是由过去的疾病或需要在图谱中单独定位的异常人口统计特征引起的。
- 如果发现异常数据集,可以考虑几个步骤。
- 首先,可以根据它们相对于图谱重点的重要性,考虑在重新整合时排除异常数据集、主体或样本(图3e)。
- 其次,可以考虑使用定制的批次协变量(图3h)、方法或参数设置(图3i)进行重新整合,从而更加注重消除异常批次效应。
- 第三,如果批次效应的来源明确,增加更多相同类型的数据库并重新整合数据可能有助于缓解批次不平衡问题。
Para_06
- 即使在去除异常值并对整合方法进行调整之后,仍然会存在一些批次效应。
- 残余的批次效应决定了细胞注释分辨率能够达到多精细的程度,因为在那之后细胞可能会根据技术效应而非生物效应分成簇。
- 因此,必须牢记这如何影响数据表示,从而影响下游分析。
Evaluation of reference quality for mapping new data
对映射新数据的参考质量进行评估
Para_01
- 由于图谱的主要用途之一是将新数据集与作为参考的图谱进行分析,因此图谱必须适合通过‘查询到参考映射’将新数据高质量地对齐到图谱(图3c)。
- 这种映射将任何未见过的单细胞数据集投影到已集成图谱的预存低维空间中,从而允许对图谱和新数据进行联合分析。
- 参考映射性能不佳可能导致对映射查询数据的错误解释。
- 解决性能不佳的问题可能需要调整集成本身,包括重新审视从数据集选择到集成超参数设置的先前步骤(图3e–i),以更好地捕获集成过程中可能存在的技术和生物学效应范围。
- 参考映射在很大程度上还取决于所使用的映射算法和底层的集成方法(‘选择最优的数据集成策略’),可能需要采用不同的集成方法以实现更好的映射效果。
Para_02
- 确定一个图谱是否适合用作参考映射需要考虑未来可能映射的数据集类型,包括技术因素的潜在差异(例如,测序协议、基因组版本)以及生物学差异(例如,来自患病供体的组织、不同的发育阶段)。
- 重要的是,来自非常不同生物学背景的数据可能会使映射复杂化(图3k),正如差异较大的数据集也会使初始图谱构建变得复杂(如‘定义重点’中所讨论的那样)。
- 为了评估图谱的参考映射潜力,可以应用‘选择最佳数据整合策略’和‘整体图谱表示质量评估’中描述的概念:在组合图谱和映射查询数据集中评估生物学特性的保留情况和批次效应校正效果。
- 可以使用多种专用指标和方法来估计映射的质量,例如,比较映射前后邻域或聚类的保留情况、通过测量不确定性指标评估细胞类型标签从参考数据到查询数据的转移置信度和准确性,以及查询细胞与参考细胞之间的距离。
Annotating the integrated atlas
标注整合图谱
Para_01
- 一旦数据成功整合,应对细胞进行重新注释,以提高注释的质量和分辨率。
- 总细胞数的增加使得可以检测到在单个数据集中可能未被注释的稀有细胞类型和状态,包括低质量液滴的群体。
- 此外,联合表示能够解决同一细胞类型在不同数据集中经常出现的矛盾注释。
- 如果在整合之前部分细胞未被标记,现在可以根据它们与整合表示中标记细胞的相似性对其进行标记。
- 重要的是,如果重新注释基于单个数据集的原始注释或多个独立专家的意见,则重新注释的图谱将成为针对特定组织达成共识注释的第一步。
Para_02
- 在此阶段,低质量的液滴(例如,空液滴和双重液滴)可能仍然存在于数据中,并应在注释之前识别出来,以与活细胞分离(图3d、j及补充注释6)。
- 正如之前讨论过的(‘数据协调与预处理’),目前尚不清楚质量控制可以在整合后完全进行,而不是在每个数据集或样本之前进行。
- 前者不仅在预处理过程中节省时间,而且通过标签转移对新映射到图谱的数据集进行自动化质量控制时,标注而非移除低质量液滴也可能是可行的。
Para_03
- 图谱的重新注释可以通过手动、自动或基于社区的众包方法完成。
- 经典且最耗费人力的方法是根据细胞在整合表示中的聚类和标记基因表达情况手动注释图谱中的所有细胞。
- 或者,可以手动或自动地将来自不同数据集的现有细胞类型标签整合到一个细胞类型层次结构中。
- 也可以通过使用标记基因或通过标签迁移来自动注释细胞。
- 最后,众包方法能够从更大规模的专家群体或网络中收集注释信息。
- 这些方法在补充注释7中进一步讨论。
Para_04
- 为了确保细胞注释的质量,必须从不同角度对其进行评估。
- 将细胞分组为细胞类型不应受到技术效应的驱动(见‘整体图谱表示质量评估’部分),例如,应避免对没有来自多个供体和数据集的细胞的聚类进行注释。
- 此外,注释标签应具有鲁棒性,通过已知标记物的表达得到验证,并与先前对单个数据集的注释大致一致,包括覆盖预期存在的细胞类型。
- 如CAP59的目标所示,引入多位生物学专家同样将提高注释的可靠性(框5)。
[div_box]
When the atlas is completed: sharing and extending the atlas
当图集完成时:分享和扩展图集
Para_01
- 最终完成的图谱将作为一个社区参考,作为资源服务于不断发展的领域,并随着新发现而持续更新。
- 为此,图谱在发布时需要向不同的用户群体开放使用。
- 此外,从长远来看,图谱需要结合新数据和信息进行扩展,以保持其时效性。
Making the atlas available to different user groups
将图集提供给不同的用户群体
Para_01
- 为了确保图谱能够作为社区资源发挥其主要作用,关键在于它们必须易于访问和重复使用。
- 这涉及两个主要要求。
- 首先,已发布的数据应有良好的文档记录。
- 这包括对所有图谱组件的描述,包含元数据协变量,并共享所有与图谱相关的代码。
- 此外,元数据协变量应尽可能遵循现有的本体命名法。
- 其次,虽然计数矩阵通常会在如 Gene Expression Omnibus (GEO)、BioStudies 和 HCA 数据门户等平台上共享,但数据也应便于不同用户群体访问。
- 为此,已经开发了专门的工具和框架。
- 对于简单的查询,例如可视化细胞中的基因表达水平或元数据类别,可以使用交互式平台。
- 对于更专业的分析,数据应易于下载,并以兼容标准数据分析平台的格式提供。
- 最后,与图谱相关的模型(例如用于查询到参考映射的模型)应公开共享,并可提供自动映射的框架。
- 值得注意的是,目前尚不清楚如何以标准化的方式共享下游图谱分析的结果,例如自定义标记列表。
- 关于图谱共享的进一步考虑在补充注释 8 中进行了详细说明。
Table 1 Different databases and platforms enable sharing of atlas data for different purposes 表1 不同的数据库和平台支持为不同目的共享图集数据

Extending and updating the atlas
扩展和更新地图集
Para_01
- 图谱可以作为不断发展的活资源,随着新数据集的出现而更新。
- 在新数据集发布时将其纳入,可以增加更多的个体或条件,从而提高元数据协变量分析的统计能力,并改善在生物条件下细胞类型和状态的覆盖范围。
- 同样,细胞注释或元数据描述符可以更新以遵循不断发展的本体论,或者包括最近研究中发现的新细胞类型。
- 重要的是,保持参考图谱的最新状态将需要相当广泛的社区和联盟的努力,就像在基因组学领域一样,标准基因组构建会迭代改进并被整个科学界使用。
Para_02
- 可以通过使用查询到参考映射算法将新数据映射到旧图谱上,从而向图谱中添加新数据。
- 当积累更多新数据时,为了捕捉或纠正新的生物学和技术变异,通过重新训练模型进行再整合将比通过查询到参考映射扩展图谱更为必要。
- 此外,参考图谱可以通过添加新的模态进行扩展,例如基于测序的单细胞转座酶可及染色质检测(ATAC-seq)和基于测序的转录组与表位的细胞索引(CITE-seq)。
- 因此,由于非转录组学模态的单细胞数据集数量增加,使用能够映射不同模态的整合模型可能很快变得至关重要。
Para_03
- 一个有趣的可能性是,通过让用户将其新数据映射到参考数据以分析自己的数据,同时在参考门户上共享其映射数据的表示,从而实现图谱扩展的规模化和流程化。
- 即使这些映射并非专门为了扩展图谱,它们也会进一步为所有用户扩展图谱。
- 这样的持续社区努力将极大地增加图谱中捕获的数据量,这种速度是单一图谱管理团队无法实现的。
[div_box]
Using integrated atlases
Para_01
- 图谱的价值来源于它所提供的生物学见解以及作为共识参考的作用(图4)。
- 图谱有潜力解答紧迫的生物医学问题,帮助理解疾病机制、开发新疗法、改进模型系统并推动疾病预后或诊断的进步。
- 此外,图谱可以用于研究整个生物体范围内的细胞功能、发育、类器官协议设计以及跨物种的进化。
- 将新数据集映射到图谱上还能够实现基于图谱的新数据分析。
- 为了促进图谱在不同领域的应用,我们在此提供了一个概述,介绍可以通过整合的图谱单独或结合新数据回答的与领域无关的生物学和技术问题。
Fig. 4: Use cases of integrated atlases.
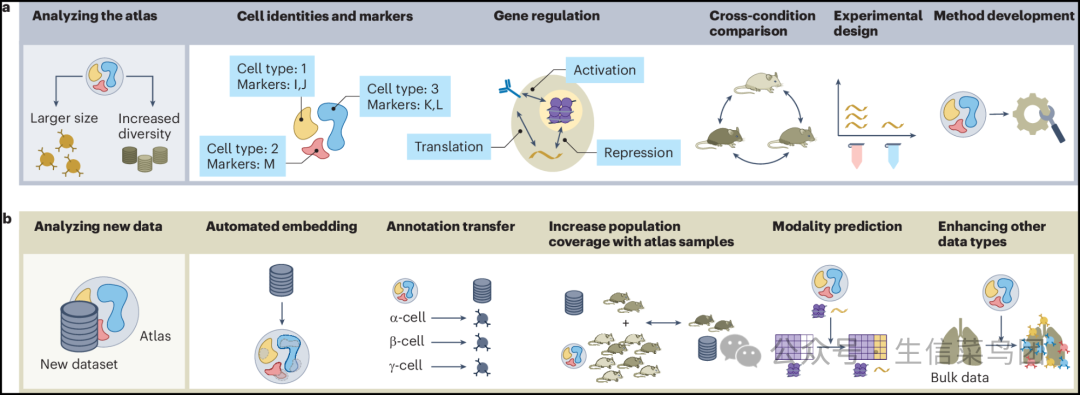
- 图片说明- 图谱中捕获的丰富信息可以通过多种方式提供新的生物学或技术见解,并且可以用作方法开发的基准(a),或者用作分析新的单细胞、空间或批量数据集的参考(b)。- 丰富的信息能够以多种方式提供新的生物学或技术洞见,既可以作为方法开发的基础(a),也可以作为分析新单细胞、空间或批量数据集的参考(b)。
Exploring the information within the atlas
探索图集中的信息
Para_01
- 标记基因、基因程序以及生物和实验技术因素对细胞类型的影响,通常会在单细胞数据集中进行研究。
- 由于图谱对生物和实验技术因素的覆盖范围更广,因此为这些分析提供了独特而全面的资源。
Cell identities and their markers
细胞身份及其标志物
Para_01
- 跨单细胞数据集的细胞类型注释很少达成一致。
- 这部分是由于细胞状态的生物学差异,但也归因于细胞类型命名法和分辨率缺乏标准化。
- 通过整合来自不同实验室、条件、解剖区域或样本处理协议的多个已注释数据集,以及关于细胞类型标签的不同专家意见,细胞图谱为建立共识细胞类型注释提供了机会。
Para_02
- 通过图谱识别的细胞类型标志物可能更具特异性、敏感性和鲁棒性,因为它们在数据集之间以及协议之间具有一致性。
- 此外,由于图谱整合了多项研究的数据,因此可以揭示在单独分析单个数据集时常常被遗漏的稀有细胞类型。
- 因此,基于图谱的标志物对于新数据集中细胞类型的注释特别有价值,并且可用于评估新发现或先前提出的标志物,以及选择用于组织染色、细胞分选和空间转录组学探针设计的标志物。
- 尽管常见的标志物鉴定策略(已在参考文献中进行了基准测试)可以应用于图谱,但由于相关聚类的数量众多及其相对层次结构,以及大量数据集和相关的批次效应,需要额外考虑一些因素。
Description of gene function and regulation
基因功能和调控的描述
Para_01
- 通过共表达分析通常可以推断单个细胞类型中的基因调控景观和分子通路。
- 这些分析得益于包含大量样本的大型异质数据集。
- 因此,图谱可以用于稳健地识别基因-基因关系和多细胞程序,后者是跨不同细胞类型共同调控的一组基因。
- 为了更好地模拟基因之间的调控关系,可以使用来自多个组学层的测量数据。
- 涵盖新兴多组学数据类型的完整组学景观的多组学图谱,可以作为不同组学层之间的桥梁。
Molecular and cellular changes across conditions
在不同条件下的分子和细胞变化
Para_01
- 为了理解表型的分子特征,例如疾病、年龄或性别,必须分析与基因表达和细胞类型组成相关的改变(以下称为‘协变量分析’)。
- 图谱在多个方面改进了协变量分析。
- 它们涵盖了大量样本和数据集,从而提高了泛化能力,并增强了检测表型与基因表达之间关联的能力。
- 这些关联还可以作为基因功能信息的额外层,超越常用的通路数据库。
- 大量的样本还能够更好地覆盖连续的临床或人口统计轨迹,例如衰老或疾病进展。
- 同样,增加的患者覆盖范围可能揭示患有相同疾病的患者之间的异质性,从而实现用于个性化医学的患者分层。
- 此外,由于图谱整合了来自多项研究的数据,因此可以将单个数据集中无法比较的生物条件结合起来。
- 例如,跨条件共享的分子特征可能有助于药物重新定位到不同疾病或组织中。
- 类似地,跨条件的差异可能有助于选择临床前模型。
- 在未来,图谱甚至可以用于基于患者的单细胞图谱构建临床分类的预测模型。
Para_02
- 图谱内的协变量分析存在多个挑战。
- 图谱中的数据集并非为了回答单一问题而生成,因此并不遵循任何特定问题的最佳实验设计。
- 此外,在构建图谱时,批次效应通常仅在整合表示中进行校正,而基因表达计数则未进行校正,这使得不同批次之间的基因表达值无法比较。
- 同样地,细胞比例分析可能会受到与批次相关的采样协议差异(例如解离技术)和组织采样位置的影响。
- 基于这些原因,图谱级别的协变量分析需要在统计模型中纳入混杂因素。
- 当生物学因素和技术因素之间存在部分混杂时,例如细胞轨迹被分割到不同数据集中,这种情况尤其具有挑战性。
- 另一种方法是针对每个数据集分别进行分析,然后合并结果。
- 此外,基于单个数据集建立的建模假设并不总是在图谱中成立。
- 例如,细胞-细胞通信工具假设所有细胞都位于同一组织中,但这对于整个图谱来说并不成立。
- 因此,标准分析方法需要根据图谱的特性进行调整。
Guiding future experimental design
指导未来实验设计
Para_01
- 图谱为改进未来实验的设计提供了多种机会。
- 例如,虽然单个数据集很少是为了评估不同技术参数如何影响数据而生成的,但图谱整合了多个数据集,使得这样的分析成为可能。
- 这可以揭示哪些技术因素需要优化,以防止细胞应激、双重事件或环境污染,或者更好地捕获特定的细胞类型。
- 此外,图谱可用于功效分析,即估计需要描绘多少细胞、样本或供体来回答特定问题。
- 这在研究稀有细胞类型时非常有用,或者在确定每细胞计数和描绘细胞数量的最佳组合用于差异基因表达分析时也很重要。
- 最后,图谱突显了当前数据中哪些细胞类型、疾病、人口统计学或其他类别尚未得到充分研究,并在未来需要更好地捕捉(框7)。
[div_box]
Developing new single-cell methods and machine-learning models
开发新的单细胞方法和机器学习模型
Para_01
- 新单细胞方法的开发高度依赖于用于方法测试和基准测试的高质量数据集。
- 高度策划的参考图谱特别适合这一用途,原因有多个。
- 首先,它们包含标准化格式的高质量数据,减少了对数据整理的需求。
- 这尤其有利于大规模通用的‘基础’模型在单细胞生物学中的开发(补充注释10)。
- 其次,它们包含多样化的大型数据,因此为方法提供了现实的挑战(例如批次效应),揭示了潜在的方法局限性。
- 第三,它们包含适用于各种基准测试任务的多样化数据。
- 这涵盖了不同的分析类型,包括跨连续协变量的轨迹推断、跨条件的差异分析以及跨批次的整合。
- 第四,由于其规模,图谱可以轻松地随机或分层分割(例如按数据集和谱系)以进行时间效率和数据复杂性的基准测试。
- 第五,图谱通常被深入研究,因此能够可靠地近似数据中存在的生物和技术效应的真实情况。
- 因此,图谱有可能作为标准的基准测试数据集,这种数据集在机器学习领域很常见,但在单细胞数据科学中仍然很少见。
[div_box]
Analyzing new single-cell, spatial or bulk data with atlases as references
以图谱为参考分析新的单细胞、空间或批量数据
Para_01
- 新数据的分析带来了许多挑战,例如数据整合和细胞类型的新注释,有时由于细胞和样本数量较少而受到限制。
- 通过利用图谱作为新数据分析的基础,可以缓解这些以及其他许多挑战。
- 图谱还可以为新数据补充额外的生物学信息,例如在跨模态表达预测、扩展对照样本库以及将批量数据与单细胞信息进行上下文关联中发挥作用。
Projecting new single-cell data into an atlas space
将新的单细胞数据投影到图谱空间中
Para_01
- 单细胞 RNA 测序(scRNA-seq)分析的核心目标之一是获得高质量、低维度的表示,从而能够识别细胞类型、状态和轨迹。
- 图谱提供了这种高质量的表示,并通过使用查询到参考的映射方法,新的数据集(查询)可以定位在图谱(参考)的表示空间中。
- 这比单独分析查询数据具有多种优势。
- 首先,稀有细胞群体在图谱中得到更好的表示,因此在映射后的查询数据集中也可以更好地识别这些细胞群体。
- 其次,图谱的表示经过优化,能够利用许多训练数据集区分生物学变异和技术变异。
- 将查询数据映射到这一表示中可以改善批次校正,特别是当查询中的批次效应与生物学变异直接混淆时,仅依靠查询数据本身无法分离这些因素。
- 第三,将查询数据映射到图谱表示空间中可以实现新数据集与图谱的快速联合分析,例如用于细胞身份注释转移和比较(更多细节参见‘注释细胞身份’和‘与对照群体的比较’部分)。
- 基于图谱的表示很可能成为未来 scRNA-seq 分析的标准步骤。
Para_02
- 查询映射到参考的成功与否取决于多个因素,包括样本特性、数据预处理以及映射方法。
- 映射后的样本应在样本生物学特征、测量特征及预处理选择方面与参考样本足够相似。
- 例如,人类参考可能无法最优地用于映射动物数据或来自其他临床前模型(如类器官)的数据。
- 这种映射可能导致在结果表示中,由于校正不足而使查询和参考之间产生过多分离,或者由于过度校正而导致不同细胞群体的合并,从而在尝试增强整合强度时出现问题。
- 在这两种情况下,整合失败都会阻碍对结果的正确解释。
- 此外,虽然可以与给定图谱一起使用的参考映射方法通常依赖于构建图谱所用的模型,但其中一些方法允许调整映射参数以适应特定上下文的需求。
- 这包括修改整合强度以及添加参考中缺失的生物相关特征(如基因)。
- 最后,尽管参考映射应始终进行评估,但目前仍缺乏支持此类评估的工具。
Para_03
- 随着捕获不同组学层的数据集数量增加,使用由单一模态(目前通常是转录组)组成的参考来分析来自不同模态的查询将变得有趣。
- 这使得各种下游分析成为可能,例如跨模态注释传递和跨组学特征相关性的识别。
- 尽管提出了多种跨组学映射策略,但它们尚未在图谱领域得到广泛应用。
Annotating cellular identities
标注细胞身份
Para_01
- 尽管手动细胞注释繁琐且容易出错,但图谱能够将高质量的参考注释自动转移到新数据集中。
- 这通常是通过将表示空间中与映射查询细胞接近的参考细胞的注释进行转移来实现的。
- 此外,由于手动设置阈值,低质量或双细胞液滴的注释往往繁琐、不可靠且不一致。
- 已对这些细胞群进行注释的图谱可以用于自动注释新数据集中的低质量液滴。
- 最后,不确定性指标可用于识别具有高不确定性的转移注释,因此这些注释更有可能是错误的。
- 研究表明,具有高不确定性标签的细胞代表了新数据中未见的细胞身份(即参考图谱中不存在的细胞身份),例如新的细胞类型或与疾病相关的细胞状态。
- 因此,图谱预计将作为未来数据集注释和分析的第一步,指导注释的手动微调。
Comparisons with a control population
与对照人群的比较
Para_01
- 基于单一数据集识别健康细胞表型与疾病特异性表型之间的差异可能会受到队列内批次效应、健康细胞群覆盖不全以及样本量小的复杂影响。
- 使用图谱作为分析新查询数据的基础可以减轻这些限制。
- 例如,将来自疾病条件的查询样本映射到健康参考图谱上可以直接识别出具有改变的、非健康表型的细胞类型。
- 图谱的规模减少了由于缺乏全面对照而导致将健康变异错误解释为疾病效应的可能性。
- 然而,仅靠图谱尚不能完全替代新数据集中的对照样本。
- 此外,使用图谱作为参考可以使我们比较图谱中包含的广泛条件下的细胞。
- 这种方法已被用于优化类器官协议或比较模型系统。
- 然而,在联合分析图谱和映射数据时必须保持谨慎,因为图谱和映射无法完全消除所有批次效应,这可能导致解释上的偏差。
Para_02
- 参考图集还可以作为评估样本质量的对照,从而避免将未预料到的技术变异误认为是生物学差异。
- 例如,如果新样本的细胞在图集表示中映射到一个意外的位置,远离相应细胞类型和生物学条件的参考细胞,这可能表明样本质量低或存在强烈的技术伪影。
Cross-modal imputation
跨模态补全
Para_01
- 为了提高对跨模态细胞状态和调控的理解,跨模态的数据填补可以丰富单模态数据集。
- 由于包含大量数据集,多模态图谱在数据填补中非常有用,从而提高了填补模型的可靠性。
- 填补技术可以在许多不同场景中使用,例如去噪、跨组学预测、空间数据中未测量基因的预测或非空间数据中空间位置的填补。
- 然而,在评估填补的可靠性时需要特别注意,特别是在填补与训练数据不密切相关的条件下。
Analysis of non-single-cell data
非单细胞数据的分析
Para_01
- 许多大规模数据集在模态和个体间可用,但缺乏对理解组织功能和疾病至关重要的细胞类型信息。
- 同样,空间数据通常无法达到单细胞分辨率。
- 为了在细胞群体水平上进行解释,可以根据单细胞数据对这些数据集进行去卷积分析。
- 图谱由于其对组织或器官中存在的细胞类型的全面覆盖以及稳健的多批次细胞类型特征,特别适合用于大规模和空间数据的去卷积分析。
Para_02
- 图谱还可以帮助更好地理解既非单细胞也非转录组的数据。
- 例如,它们可以帮助识别可能受干扰影响的细胞类型,如全基因组关联研究中与疾病相关的基因组变异所影响的细胞类型,以及可能被特定药物靶向的细胞类型。
- 最终,当收集到足够的匹配单细胞和临床数据时,可能会开发出能够从图像或其他临床测量中推断细胞水平特征(如细胞比例)的模型。
[div_box]
Conclusion and outlook
Para_01
- 随着数据整合方法的成熟和单细胞数据集的广泛可用,绘制图谱的研究正变得越来越普遍。
- 图谱资源有望在各研究群体间建立共识,并对生物医学研究产生影响。
- 然而,构建图谱的标准尚不完善,图谱的应用场景仍在探索中。
- 在本综述中,我们讨论了构建和使用图谱时需要考虑的因素及机遇,以启动对该领域标准的讨论。
Para_02
- 在图谱构建领域,仍然存在许多可以通过基准测试获益的开放性问题,并且需要新的数据集、技术和方法。
- 首先,缺乏对不同图谱构建流程的系统性比较。
- 其次,随着图谱变得更加全面和复杂,包括跨物种、纵向、全生物体和多模态数据,对于能够适应这些复杂场景的整合方法的需求将会增加。
- 机器学习领域的最新进展,例如能够为大型和多样化数据集生成广泛可用表示的基础模型,在单细胞领域也可能具有实用性。
- 第三,为了实现群体范围和跨组学的图谱构建,并推广其在临床中的应用,需要降低单细胞分析技术的成本和劳动强度。
- 这些开放性问题和潜在解决方案在补充注释 11 中进一步讨论。
Para_03
- 同样,鉴于图谱被设计为社区资源且其可用性至关重要,我们确定了提升可用性的关键领域。
- 首先,交互界面和标准分析流程的性能会随着数据集规模的增大而下降。
- 为了克服这一问题,更广泛地采用图形处理单元(GPU)加速工具、开发更紧凑的数据表示方法(如将细胞压缩成元细胞、将数据编码为基础模型或更简单的生成模型)或将细胞和基因景观以标准化的人类或机器可读形式进行描述将会是有益的。
- 其次,随着图谱复杂性的增加,其可视化和解释的难度也随之提高。
- 因此,应采用工作流来简化跨越组织分辨率和组学层次的图谱的解释和可视化。
- 第三,基于参考图谱映射新数据的现有分析工作流仍处于原型阶段,需要进一步开发和测试。
- 第四,需要涵盖代表性不足的供体群体(如特定祖先群体)的单细胞数据集,以使图谱更具通用性和鲁棒性。
- 第五,尽管图谱在分子生物学、医学和计算科学等领域具有巨大潜力,但目前的访问接口主要针对生物信息学社区。
- 因此,有必要进一步开发针对不同用户需求的数据访问选项,包括交互平台和应用程序编程接口访问点。
Para_04
- 随着新的单细胞数据集生成,图谱的规模和复杂性也将增长。
- 这将带来关于最佳图谱规模以及图谱何时可以被视为‘完整’的问题。
- 未来的研究需要系统地评估在什么情况下增加更多数据不再提高生物信息(例如细胞状态和谱系)的覆盖范围或整合的质量。
- 目前,如何在实践中确定最佳图谱规模仍然不清楚。
- 部分原因是图谱研究的目标具有多样性。
- 健康细胞类型的变异,包括稀有细胞状态,可能通过当前可用的数据集进行全面检索。
- 相比之下,要全面涵盖不同人群和条件下的遗传和表型多样性,则需要大量的样本,这在短期内不太可能实现。
Para_05
- 尽管参考图谱有诸多承诺,它们也存在局限性。
- 首先,图谱依赖于整合技术以去除数据集之间的批次效应。
- 然而,这种方法很少能完美实现,并且当批次效应较强时,还会移除生物变异。
- 这可能会限制可检索细胞群体的分辨率。
- 其次,就像任何单细胞数据集一样,图谱的设计都有特定的目标,因此可能不适合回答某些生物学问题。
- 例如,如果图谱构建者专注于提供健康参考,这可能会限制基于图谱对其他条件数据的分析。
- 第三,图谱构建需要大量的人力和计算资源,随着图谱规模的扩大,这种需求可能会增加。
- 因此,近期的研究提出通过自动化管道补充高质量参考图谱,这些管道能够针对具体生物学问题进行更模块化和快速的数据整合。
- 第四,图谱的质量及其长期维护因缺乏最佳实践标准而参差不齐。
- 在本综述中,我们旨在迈出第一步,以建立这些标准。
Para_06
- 虽然图谱预计将在医学领域产生深远影响,从疾病靶点识别和毒性预测到在诊所的直接应用,但医学并不是唯一一个预计将被图谱改变的领域。
- 例如,跨物种图谱可能为系统发育和生态位提供见解。
- 同样,生态学和农业图谱可以整合跨区域的数据集,揭示环境与生物体之间的相互作用。
- 然而,在这样的图谱能够创建之前,这些领域必须生成更多的单细胞数据集。
- 因此,在未来,非生物医学领域的图谱可能会集中于模式生物,因为这些生物有足够的数据和社区关注。
Para_07
- 在这篇综述中,我们概述了构建、使用和共享图谱的当前考虑因素和建议,并强调了这些过程中值得进一步研究和发展的方面。
- 我们预计这里收集的见解将有助于为图谱构建设定共同标准,并推动图谱在单细胞研究中的广泛应用。
- 共同作用下,这将为描述细胞生物学的共识方法铺平道路,增强图谱对分子生物学和医学的影响。
Data availability
Para_01
- 已发布的 scRNA-seq 数据集的最终分析结果汇总在补充表 2 中,中间结果可从 https://github.com/lueckenlab/single-cell-papers-trends/ 获取。
Code availability
Para_01
- 图1所示已发表的单细胞RNA测序数据集的分析代码可在https://github.com/lueckenlab/single-cell-papers-trends/获取。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-02-13,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号