SCENIC+效果不好?新的GRN算法 LINGER | Nat.Biotechnol |

SCENIC+效果不好?新的GRN算法 LINGER | Nat.Biotechnol |


Basic Information
- 英文标题:Inferring gene regulatory networks from single-cell multiome data using atlas-scale external data
- 中文标题:使用图谱规模的外部数据从单细胞多组学数据推断基因调控网络
- 发表日期:12 April 2024
- 文章类型:Article
- 所属期刊:Nature Biotechnology
- 文章作者:Qiuyue Yuan | Zhana Duren
- 文章链接:https://www.nature.com/articles/s41587-024-02182-7
Abstract
Para_01
- 现有的基因调控网络(GRN)推断方法依赖于单一的基因表达数据或较低分辨率的批量数据。
- 尽管最近整合了染色质可及性和RNA测序数据,但从有限的独立数据点中学习复杂机制仍然是一个巨大的挑战。
- 在此,我们介绍了LINGER(用于基因调控的终身神经网络),这是一种从单细胞配对的基因表达和染色质可及性数据中推断GRNs的机器学习方法。
- LINGER结合了跨不同细胞环境的大规模外部批量数据以及转录因子基序的先验知识作为流形正则化。
- 与现有方法相比,LINGER实现了四倍到七倍的相对准确性提升,并揭示了全基因组关联研究的复杂调控景观,从而增强了对疾病相关变异和基因的解释能力。
- 在从参考单细胞多组学数据推断GRN之后,LINGER能够仅从批量或单细胞基因表达数据估计转录因子活性,利用丰富的可用基因表达数据从病例对照研究中识别驱动调节因子。
Main
Para_01
- GRNs 是分子调节因子的集合,这些调节因子相互作用并在特定的细胞环境中决定基因的激活和沉默。
- 全面理解基因调控对于解释细胞如何执行多种功能、细胞如何根据不同环境改变基因表达以及非编码遗传变异如何导致疾病至关重要。
- GRNs 由转录因子(TFs)组成,这些转录因子结合 DNA 调控元件以激活或抑制目标基因的表达。
Para_02
- 推断基因调控网络(GRNs)是核心问题,已经有许多尝试来解决这个问题。
- 基于共表达的方法,如 WGCNA、ARACNe 和 GENIE3 通过捕捉转录因子(TF)与靶基因(TG)的共变性,从基因表达中推断 TF 对 TG 的反式调控。
- 这些网络具有无向边,无法区分 TFA 到 TFB 的方向。
- 此外,共表达被解释为相关性而不是因果调控。
- 全基因组范围的染色质可及性测量,如 DNase-seq 和转座酶可及染色质测序(ATAC-seq),可以定位调控元件(REs),通过基序匹配将 TF 与 RE 连接,并将 RE 与其附近的 TG 连接。
- 然而,TF 足迹方法无法区分共享基序的同一家族内的 TF。
- 为了解决这一局限,我们开发了一个统计模型 PECA,通过在多种细胞类型中拟合 TF 表达和 RE 可及性来预测 TG 表达。
- 然而,这个问题仍未完全解决,因为批量数据中细胞类型的异质性限制了推断的准确性。
Para_03
- 单细胞测序技术的出现使得在单个细胞类型水平上进行高度精确的调控分析成为可能。
- 单细胞 RNA 测序(scRNA-seq)数据通过共表达分析,如 PIDC 和 SCENIC,可以推断特定细胞类型的转调控关系。
- 用于转座酶可及染色质的单细胞测序(scATAC-seq)可以通过 DeepTFni 等方法推断转调控关系。
- 许多方法整合了未配对的 scRNA-seq 和 scATAC-seq 数据以推断转调控关系。
- 这些方法包括 IReNA、SOMatic、UnpairReg、CoupledNMF 和 DC3 等,通过基序匹配将转录因子与调控元件联系起来,并使用调控元件与靶基因之间的协变或物理碱基对距离将其链接到靶基因。
- 最近,scJoint 被开发用于将标签从 scRNA-seq 数据转移到 scATAC-seq 数据,这可能有助于改进细胞基因调控网络的推断。
- 尽管付出了大量努力,基因调控网络推断的准确性仍然令人失望地低,仅略高于随机预测。
Para_04
- 单细胞测序的最新进展为解决这些挑战提供了机会,例如 SCENIC+。
- 然而,在基因调控网络推断中仍然存在三个主要挑战。
- 首先,从有限的数据点学习这种复杂的机制仍然是一个挑战。
- 虽然单细胞数据提供了大量细胞,但大多数细胞并不是独立的。
- 其次,将先验知识(如基序匹配)纳入非线性模型具有挑战性。
- 第三,通过实验数据评估的推断出的基因调控网络准确性仅略好于随机预测。
Para_05
- 为克服这些挑战,我们提出了一种称为 LINGER(基因调控的终身神经网络)的方法。
- 本文在基因调控网络推断领域做出了多方面的贡献。
- 首先,LINGER 使用了终身学习,这是一种先前定义的概念,它结合了大规模外部批量数据,缓解了数据有限但参数广泛的问题。
- 其次,LINGER 通过流形正则化整合了转录因子-反应元件基序匹配知识,使先验知识能够融入模型中。
- 第三,LINGER 的准确性提高了四倍到七倍。
- 第四,LINGER 能够仅从基因表达数据中估计转录因子活性,识别驱动调节因子。
Results
LINGER: using bulk data to infer GRNs from single-cell multiome data
使用批量数据从单细胞多组学数据推断基因调控网络(GRNs)
Para_01
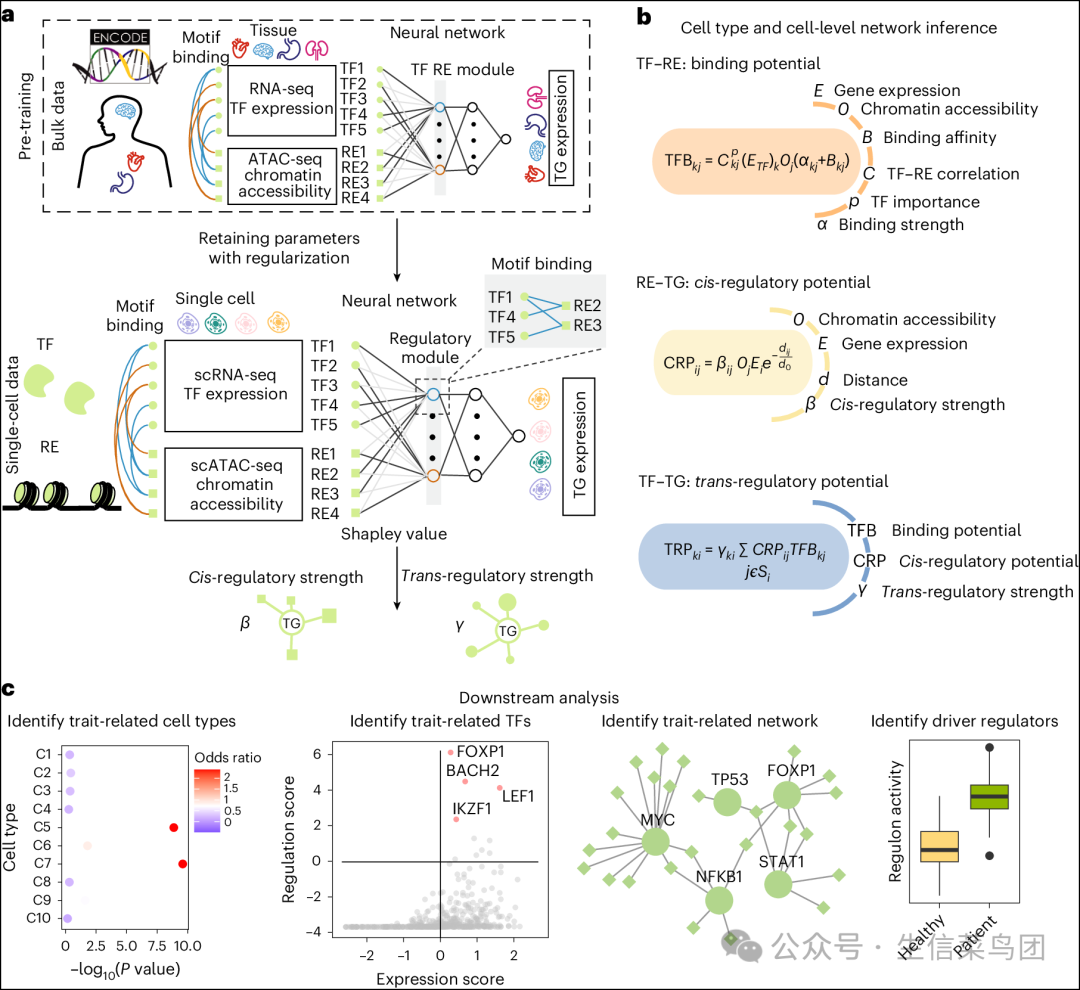
- LINGER 是一个计算框架,旨在从单细胞多组学数据推断基因调控网络(图 1 和方法)。
- 它使用基因表达和染色质可及性的计数矩阵以及细胞类型注释作为输入,提供细胞群体 GRN、细胞类型特异性 GRN 和细胞水平 GRN。
- 每个 GRN 包含三种类型的相互作用,即转调控(TF–TG)、顺调控(RE–TG)和转录因子结合(TF–RE)。
- 请注意,TF–TF 相互作用包含在 TF–TG 对中,但 TF 自我调控由于缺乏额外数据难以建模,因此未考虑。
- LINGER 的特点是能够整合来自外部批量数据的全面基因调控谱型。
- 这是通过终身机器学习,也称为连续学习实现的。
- 终身学习的概念是,过去学到的知识可以帮助我们用少量数据或努力学习新事物。
- 终身学习已被证明可以利用之前任务中学到的知识来更好地学习新任务。
Fig. 1: Schematic overview of LINGER.
错误!!! Request timed out. 错误!!! Request timed out. 错误!!! Request timed out.
LINGER improves the cellular population GRN inference
LINGER 改进了细胞群体基因调控网络的推断
Para_01
- 为了评估 LINGER 的性能,我们使用了来自 10× Genomics 的外周血单核细胞(PBMCs)的公开多组学数据集(详见方法部分)。
- 为了研究线性模型是否足以建模基因表达或是否需要非线性模型,我们在这两个模型之间进行了比较分析。
- 第一个模型采用弹性网络预测转录因子和调控元件对目标基因的表达。
- 第二个模型是单细胞神经网络(scNN),它是一个三层神经网络模型,与 LINGER 具有相同的架构。
- 我们使用五折交叉验证评估了这两个模型的基因表达预测能力。
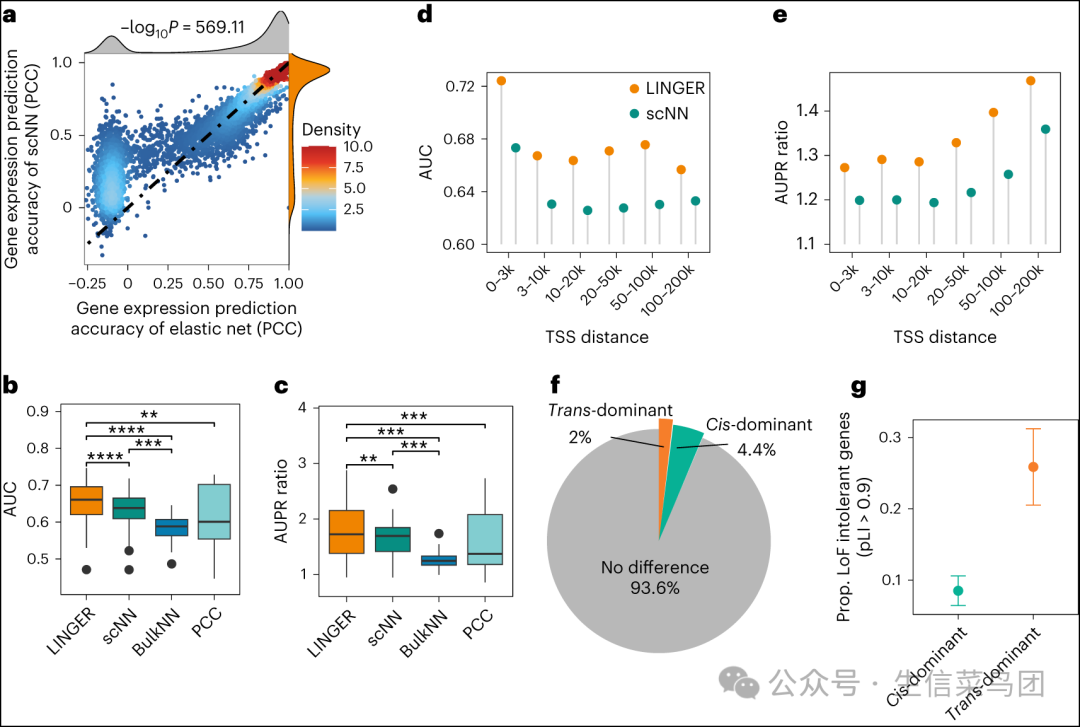
- 我们发现 scNN 在预测基因表达方面优于弹性网络,尤其是在那些在弹性网络预测中表现出负皮尔逊相关系数(PCC)的基因中,差异更为显著(−log10P = 572.09;图 2a)。
- 这表明三层神经网络模型 scNN 在预测基因表达方面优于弹性网络模型。
Fig. 2: LINGER improves the cellular population GRN inference.
- 图片说明- 预测基因表达与真实基因表达之间的相关性,显示 scNN 比弹性网具有更高的准确性。x 轴表示弹性网预测的基因和实际基因表达在细胞间的皮尔逊相关系数(PCC),而 y 轴给出 scNN 的 PCC。点代表基因,点的颜色表示密度。- 图 b 至 e 中颜色分布表示不同方法:橙色为 LINGER;灰色为弹性网;深绿色为 scNN;蓝色为 BulkNN;浅蓝色为 PCC。零假设检验结果得到 t 统计量的效果大小为 53.46,自由度 df = 15,659,-log10P = 572.09 和 95% 置信区间 [0.058, 0.063],来自双侧配对 t 检验。- 图 b 显示了所有真实数据中预测的转调控强度的性能指标 AUC 的箱线图。b 和 c 的真实数据是来自血液细胞的 20 个 ChIP-seq 数据点中的转录因子目标(n = 20 独立样本)。PCC 表示调节元件(RE)的染色质可及性与靶基因(TG)表达之间的皮尔逊相关系数。- 请注意,本研究中的所有箱线图表示最小值和最大值,即不被视为异常值的最小和最大值;中心为中位数;箱子的边界为第 25 百分位数(Q1)到第 75 百分位数(Q3);须为 1.5 倍的(Q3 - Q1)。- 本研究使用以下符号表示统计显著性:ns,P > 0.05;P ≤ 0.05;P ≤ 0.01;P ≤ 0.001;****P ≤ 0.0001。当显示显著性水平时,我们隐藏 ns 符号。具体来说,LINGER 和 scNN 的 P 值为 8.32 × 10^-6,LINGER 和 BulkNN 的 P 值为 8.57 × 10^-5,LINGER 和 PCC 的 P 值为 1.24 × 10^-3。- 图 c 显示了预测的转调控强度的性能指标 AUPR 比率的箱线图。b 和 c 的 P 值来自单侧配对 t 检验。具体来说,LINGER 和 scNN 的 P 值为 3.49 × 10^-3,LINGER 和 BulkNN 的 P 值为 2.13 × 10^-4,LINGER 和 PCC 的 P 值为 4.53 × 10^-4。- 图 d 显示了 LINGER 推断的顺调控强度的 AUC。d 和 e 的真实数据是来自 eQTLGen 的变异-基因关联。我们根据调节元件(RE)与靶基因转录起始位点(TSS)的距离将 RE-TG 对分为不同的组。- 图 e 显示了顺调控强度的 AUPR 比率。- 图 f 显示了转主导或顺主导基因的分类。转录因子(TFs)对预测转主导基因的表达贡献更大,而调节元件(REs)对顺主导基因的贡献更大。- 图 g 显示了转主导和顺主导基因成为功能丧失(LoF)耐受基因的概率。点表示从二项分布估计的成功概率,分别为 0.26 和 0.09。转主导基因的独立样本量 n = 317,顺主导基因的独立样本量 n = 693。数据显示为均值 ± 1.96 × 标准差。
Para_02
- 为了展示整合外部批量数据的实用性和有效性,我们将 LINGER 与 scNN、BulkNN 和 PCC 进行了比较。
- 为了评估转调控强度的性能,我们从染色质免疫沉淀测序(ChIP–seq)数据中收集了转录因子的假定靶标,并使用系统标准(方法)获得了血液细胞中的 20 个数据集作为真实数据(补充表 1)。
- 对于每个真实数据,我们通过滑动转调控预测计算了接收者操作特征曲线下面积(AUC)和精确率-召回率曲线下面积(AUPR)比值(见方法)。
- 结果显示,scNN 的表现优于 PCC 和 BulkNN。
- 与其他方法相比,LINGER 表现更好,在所有真实数据中具有显著更高的 AUC(图 2b)和 AUPR 比值(图 2c)。
Para_03
- 为了验证 LINGER 的顺式调控推断,我们计算了顺式调控系数与表达数量性状位点(eQTL)研究的一致性,这些研究将基因型变异与其目标基因联系起来。
- 我们从 GTEx 和 eQTLGen 下载了由 eQTL 在全血中定义的变异-基因链接作为真实数据集。
- 由于调控元件与目标基因之间的距离对预测非常重要,我们将调控元件-目标基因对分为不同的距离组。
- 在 eQTLGen 中,LINGER 在所有不同距离组中的 AUC 和 AUPR 比率均高于 scNN(图 2d,e),在 GTEx 中也是如此(扩展数据图 1a,b)。
- 上述结果表明,通过利用外部数据,LINGER 提高了顺式调控和反式调控强度的推断。
Para_04
- 接下来,我们试图研究基因的主要调控机制;即,一个基因主要由顺式调控还是反式调控所主导。
- 为了回答这个问题,我们通过双侧非配对 t 检验比较了顺式调控和反式调控的 Shapley 值的平均值,并进行了 Bonferroni P 值校正。
- 我们的研究结果表明,大多数基因在顺式调控和反式调控的主导性上没有显著差异。
- 具体来说,4.37% 的基因是顺式调控占主导,而 2.00% 的基因是反式调控占主导(图 2f)。
- 为了区分反式调控占主导和顺式调控占主导的基因之间的进化差异,我们使用 pLI(‘丧失功能不耐受概率’的估计值)比较了它们的选择压力。
- 我们观察到,在反式调控占主导的基因组中,具有高 pLI (>0.9) 的选择受限基因的比例大约是顺式调控占主导基因组的三倍(图 2g)。
- 之前的研究发现,与疾病相关的基因在 GWAS 中富集于选择受限基因,而 eQTL 基因在选择受限基因中则较少见。
- 这些观察结果突显了反式调控网络在理解复杂疾病中的重要性。
- 功能富集分析显示,顺式调控占主导的基因在 38 个 GTEx 衰老特征中显著富集(补充表 3),这与结论一致,即染色质可及性变化发生在年龄相关性黄斑变性中。
Para_05
- 为了理解参数敏感性,我们系统地评估了转录因子-反应元件(TF–RE)基序匹配、顺式反应元件(cis-REs)与转录起始位点(TSS)的距离、激活函数、隐藏层中的节点数以及元细胞生成方法对单细胞神经网络(scNN)的影响。
- 注意,sigmoid 激活函数不会改善基因表达预测,但会改善基因调控网络(GRN)推断。
- 通过设置权重,使用流形正则化损失正确利用基序匹配信息可以提高性能。
- 与权重为 0 相比,权重为 0.01 在 AUC 和 AUPR 比率上分别将性能提高了 100% 和 80%。
- 权重为 10 的性能相比 0.01 有所下降。
- 为了验证我们方法对替代元细胞生成方法的鲁棒性(参见方法中的‘PBMC 10× 数据’),我们使用 SEACells 生成的元细胞代替我们原来的元细胞。
- SEACells 元细胞与我们原来的元细胞之间没有显著的性能差异(双侧配对 t 检验,P = 0.89)。
- 在 200 kb、500 kb、1 Mb 和 2 Mb 中,使用 1 Mb 范围内的反应元件表现最佳。
Para_06
- 我们通过利用额外的数据源评估了 LINGER 的终身学习能力。
- 我们将 ENCODE 数据分为两批(ENCODE1 和 ENCODE2),并进行了两轮预训练,然后在 PBMCs 单细胞多组学数据上进行了训练(ENCODE1+ENCODE2+sc)。
- 我们将其结果与仅使用一批 ENCODE 数据进行预训练的结果(ENCODE1+sc)进行了比较。
- 扩展数据图 2h 显示,与单次预训练相比,增加第二轮预训练使基于 AUC 比率的 85.5%(20 个中的 17 个)和基于 AUPR 比率的 75%(20 个中的 15 个)的 ChIP–seq 数据的 TF–TG 推断性能得到了提升。
- 这验证了 LINGER 通过从不同数据集中进行增量学习来不断改进的能力。
LINGER improves the cell type-specific GRN inference
LINGER 提高了细胞类型特异性基因调控网络的推断
Para_01
- 我们评估了LINGER在PBMCs单细胞多组学数据中以及来自单核染色质可及性和mRNA表达测序(SNARE-seq)数据的H1、BJ、GM12878和K562细胞系的体外混合中的细胞类型特异性基因调控网络推断(方法)。
- 为了评估转录因子与调控元件结合预测,我们使用ChIP-seq数据作为真实情况,包括血液中四种细胞类型的20个转录因子和H1细胞系的33个转录因子。
- 来自ChIP-seq数据的潜在靶标作为转录调控潜力的真实情况。
- 对于顺式调控潜力,我们整合了三种主要血液细胞类型的启动子捕获Hi-C数据以及六种免疫细胞类型的单细胞eQTL数据,作为PBMCs的真实情况。
Para_02
- 为了评估转录因子(TF)与调控元件(RE)的结合潜力,我们将我们的方法与转录因子-调控元件相关性(PCC)和基序结合亲和力进行了比较。
- 例如,在幼稚 CD4 T 细胞中,LINGER 对于 ETS1 的曲线下面积(AUC)达到了 0.92,精确率-召回率曲线下面积(AUPR)比值为 5.17,这比 PCC(AUC 为 0.78;AUPR 比值为 2.71)和基序结合亲和力(AUC 为 0.70;AUPR 比值为 1.92)有所提高(图 3a,e)。
- 对于 H1 细胞系中的 MYC 结合位点,LINGER 的表现优于基于 PCC 和基序结合亲和力的预测(扩展数据图 3a,b)。
- 对于 PBMCs 中的所有 20 个转录因子,LINGER 一直表现出最高的 AUC 和 AUPR 比值,并且整体分布显著高于其他方法在 PBMCs 中的表现(P ≤ 8.72 × 10−5;图 3b,c 和补充表 6)。
- LINGER 在 H1 数据上的表现也优于其他方法(P ≤ 6.68 × 10−6;扩展数据图 3c,d)。
- 此外,我们将 LINGER 与一种最先进的方法 SCENIC+ 进行了比较,该方法从多组学单细胞数据中预测转录因子-调控元件对。
- 鉴于 SCENIC+ 不为所有调控元件提供连续评分,我们使用 F1 分数作为准确性的衡量标准。
- 图 3d 显示,LINGER 在所有 20 个转录因子结合位点预测中的表现更好。
Fig. 3: Systematic benchmarking of cell type-specific TF–RE binding potential and cis-regulatory potential performance.

- 图片说明- a,e,ETS1在初始CD4 T细胞中结合潜力的受试者工作特征曲线和精确率-召回率曲线。a和e的真实数据是初始CD4+ T细胞中ETS1的ChIP–seq数据。a到e中的颜色代表用于预测转录因子-调控元件调控关系的不同方法。橙色表示LINGER;绿色表示转录因子表达与调控元件染色质可及性之间的皮尔逊相关系数(PCC);蓝色表示转录因子对调控元件的基序结合亲和力。- b,c,不同转录因子和细胞类型中结合潜力的AUC和AUPR比率的小提琴图。真实数据来自血液中不同细胞类型的20个转录因子的ChIP–seq数据。原始数据见补充表6。b中,比较LINGER与PCC和结合的AUC的一侧配对t检验得到的t统计量分别为8.99和18.25,自由度为19,p值分别为1.42×10^-8和8.34×10^-14,95%置信区间分别为[0.17, Inf]和[0.17, Inf]。c中,比较LINGER与PCC和结合的AUPR比率的一侧配对t检验得到的t统计量分别为4.65和5.44,自由度为19,p值分别为8.72×10^-5和1.49×10^-5,95%置信区间分别为[1.31, Inf]和[1.51, Inf]。- d,结合潜力的表现指标F1分数。每个点代表真实数据(n = 20个独立样本)。d、h和k的p值基于一侧配对t检验。- f,g,初始CD4+细胞中顺式调控潜力的AUC和AUPR比率。f到h的真实数据是启动子捕获Hi-C数据。RE-TG对被分为六个距离组,范围从0-5 kb到100-200 kb。PCC计算的是TG表达与RE染色质可及性之间的相关性。距离表示到TSS的距离衰减函数。随机表示从0到1的均匀分布。- h,LINGER和SCENIC+在初始CD4+细胞中顺式调控的F1分数(n = 9个独立样本)。- i,j,顺式调控潜力的AUC和AUPR比率。真实数据来自六种免疫细胞类型的eQTL数据。- k,初始B细胞中顺式调控潜力的F1分数。真实数据来自初始B细胞的eQTL数据(n = 9个独立样本)。该图对应于PBMC数据。
Para_03
- 为了评估顺式调控潜力,我们将 LINGER 与四种基线方法进行了比较,包括基于距离的方法、RE–TG 相关性(PCC)、随机预测和 SCENIC+。
- 我们将 Hi-C 数据的 RE–TG 对分为六个距离组,范围从 0–5 kb 到 100–200 kb。
- 在幼稚 CD4 T 细胞中,LINGER 在所有距离组中的 AUC 范围为 0.66 至 0.70,AUPR 比率为 1.81 至 2.16,而其他方法接近随机水平。
- 在其他细胞类型中,LINGER 也表现出对基线方法的一致优越性。
- 由于可用于分组的距离对数量不足,所有 eQTL 对都被视为正标签。
- 在所有细胞类型中,LINGER 的 AUC 和 AUPR 比率均高于基线方法。
- 我们还将我们的方法与 SCENIC+ 进行了比较,后者输出预测的 RE–TG 对但不提供重要性评分。
- 我们选择了相同数量的最高排名的 RE–TG 对,并使用对应于第 10 百分位到第 90 百分位的九个阈值计算了 F1 分数。
- 结果表明,基于 Hi-C 数据,LINGER 在所有细胞类型中的 F1 分数显著高于 SCENIC+。
- 以 eQTL 为真实标签,LINGER 的 F1 分数显著高于 SCENIC+ 以及其他细胞类型。
Para_04
- 为了评估转调控潜力的准确性,我们选择了 GENIE3 和 PIDC 进行比较,基于之前工作中选择的单细胞数据 GRN 推断基准文献(见方法)。
- 此外,我们将 LINGER 与 PCC 和 SCENIC+ 进行了比较。
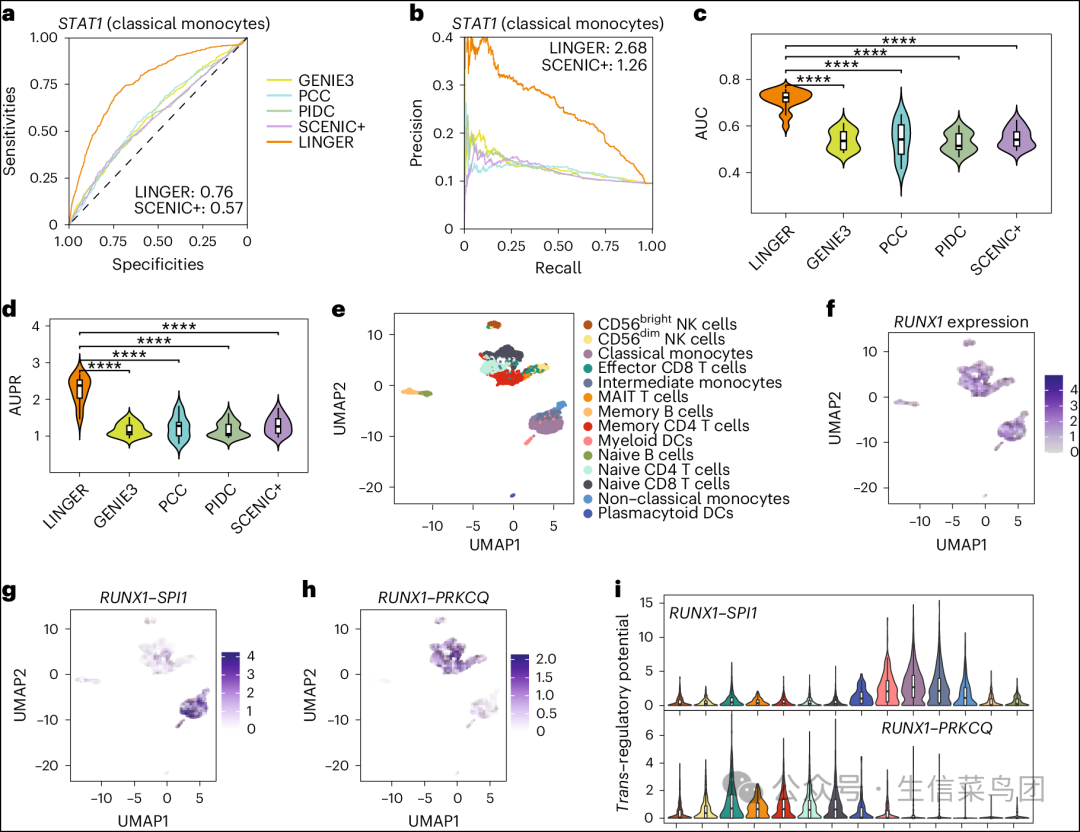
- 对于经典单核细胞中的 STAT1,LINGER 提高了预测性能,如 AUC 为 0.76 对比 0.57–0.59,AUPR 比率为 2.60 对比 1.26–1.36(图 4a,b)。
- 在 H1 中的 CTCF 也观察到了类似的改进(扩展数据图 3p,q)。
- 其他方法在真实数据集上的平均 AUPR 比率是 1.17–1.29,比随机预测高出 0.17–0.29 个单位,而 LINGER 达到了比随机预测高出 1.25 个单位,表明相对增加了四到七倍(图 4d)。
- 总体而言,LINGER 在所有 20 种 PBMCs 转录因子中始终优于其他方法,具有显著更高的 AUC 和 AUPR 比率(P ≤ 9.49 × 10−9;图 4c,d 和补充表 7)。
- LINGER 在 H1 细胞系中也优于其他竞争对手(P ≤ 3.00 × 10−8;扩展数据图 3r)。
- 与仅使用 scRNA-seq 数据的 GENIE3 和 PIDC 不同,我们的方法通过整合 scRNA-seq 和 scATAC-seq 有效将细胞数据量增加了一倍。
- 为了更公平的比较,我们移除了预训练并仅使用一半数量的细胞作为输入(scNN_half)。
- 与其他竞争对手相比,scNN_half 仍然显著优于所有其他方法(扩展数据图 2b)。
- 我们还评估了细胞类型特异性的转调控潜力,以预测 TF 扰动下的直接差异表达基因(DEGs),使用扰动实验数据作为真实数据。
- 我们从 KnockTF 数据库收集了八个 PBMCs 数据集(补充表 8)。
- 扩展数据图 4a,b 显示 LINGER 优于所有其他方法(P ≤ 3.72 × 10−4)。
Fig. 4: Systematic benchmarking of cell type-specific trans-regulatory potential performance.
- 图片说明- a,b,经典单核细胞中 STAT1 转调控潜力推断的受试者工作特征曲线和精确率-召回率曲线。这部分的真实数据来自 PBMCs 中相应细胞类型的 ChIP-seq 数据中的转录因子潜在目标。- c,d,不同转录因子和细胞类型中转调控潜力性能的 AUC 和 AUPR 比值的小提琴图。原始数据见补充表 7。单侧配对 t 检验的样本量为 20。- 对于 c,GENIE3、PCC、PIDC 和 SCENIC+ 的 -log10(P 值) 分别为 11.12、7.72、11.13 和 10.17。- 对于 d,相应的 -log10(P 值) 分别为 9.59、8.02、9.22 和 8.47。- e,包括 14 种细胞类型的 PBMCs 的均匀流形逼近和投影 (UMAP) 图。NK 细胞指自然杀伤细胞;MAIT 指黏膜相关不变 T 细胞;DCs 指树突状细胞。- f,PBMCs 中 RUNX1 表达的 UMAP 图。- g,PBMCs 中 RUNX1(转录因子)–SPI1(靶基因)细胞水平转调控潜力的 UMAP 图。- h,PBMCs 中 RUNX1(转录因子)–PRKCQ(靶基因)细胞水平转调控潜力的 UMAP 图。- i,不同细胞类型的细胞水平转调控潜力的小提琴图。每个箱线图的样本量是每种细胞类型的细胞数量,范围从 98 到 1,848。该图对应于 PBMCs。
Para_05
- 构建单细胞水平基因调控网络(GRN)的理由与构建细胞类型特异性GRN的理由相同,只是将细胞类型特异性术语替换为单细胞术语(方法)。
- 我们以RUNX1为例展示了转调控的结果。
- RUNX1对于建立确定性造血至关重要,并在几乎所有外周血单核细胞类型中高表达(图4e,f)。
- RUNX1在单核细胞(经典、非经典和中间型)和髓系树突状细胞中调节SPI1(图4g,i),而在CD56dim自然杀伤细胞、效应CD8 T细胞、黏膜相关不变T细胞、记忆CD4 T细胞、幼稚CD4 T细胞和幼稚CD8 T细胞中调节PRKCQ(图4h,i)。
- 这个例子说明了LINGER在单细胞水平上可视化基因调控的能力。
LINGER reveals the regulatory landscape of GWAS traits
LINGER揭示了GWAS性状的调控景观
Para_01
- 全基因组关联研究已经确定了数千个疾病变异,但涉及变异调控基因的活跃细胞和功能仍然 largely unknown。我们整合了全基因组关联研究的汇总统计数据和特定细胞类型的基因调控网络,以识别相关的细胞类型、关键转录因子和子网络(方法)。
- 我们为每个细胞类型的转录因子定义了一个性状调控评分,用于衡量 GWAS 基因在转录因子下游的富集程度。
- 在与性状相关的细胞类型中,具有高性状调控评分的转录因子应该表达以执行其功能。
- 我们通过评估转录因子表达与性状调控评分之间的一致性来识别与性状相关的细胞类型。
Para_02
- 在我们对炎症性肠病(IBD)的特定研究中,我们基于来自 NHGRI-EBI GWAS 目录的大约 330,000 人的 GWAS 元分析收集了风险位点,用于研究 GCST90225550。
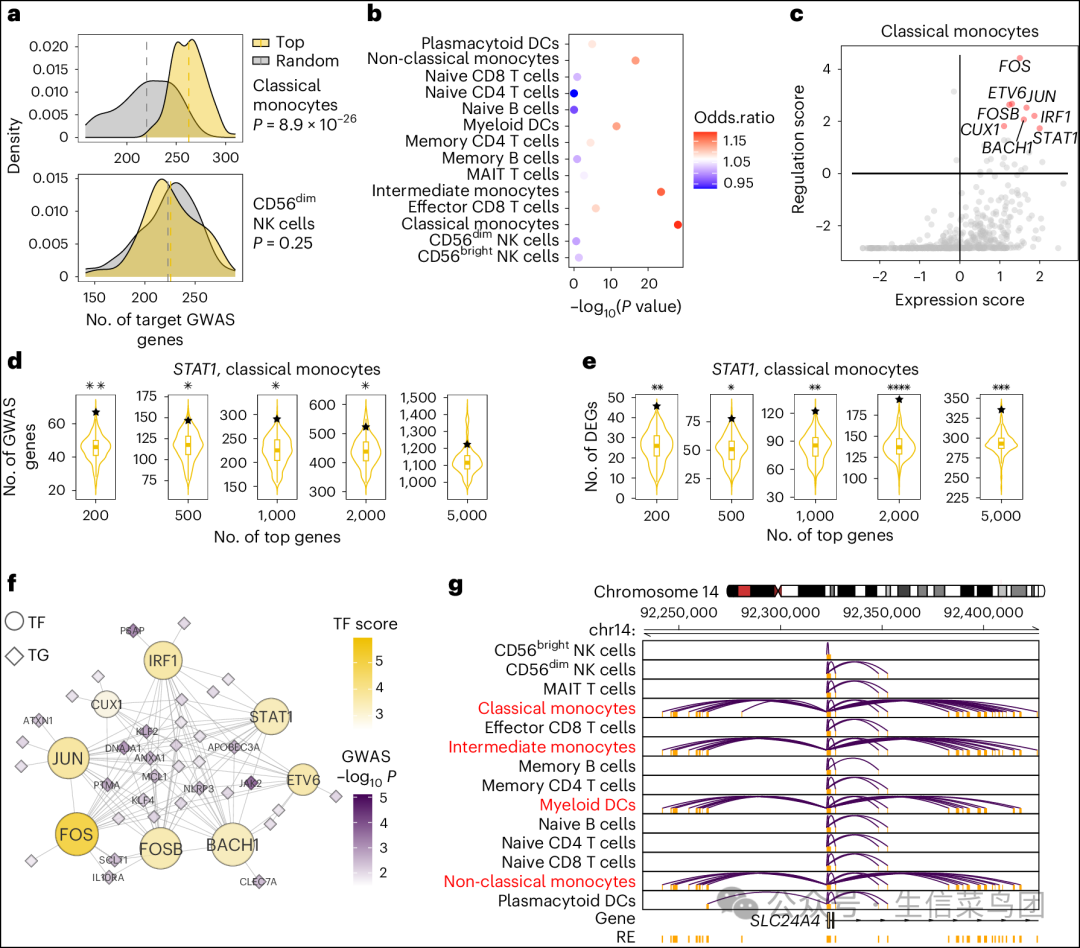
- 图 5a 显示,在经典单核细胞中,最高表达的转录因子的特征调控得分显著高于随机选择的转录因子(P = 8.9 × 10^-29,单侧非配对 t 检验),而对于不相关的细胞类型如 CD56dim 自然杀伤细胞则没有显著差异。
- 外周血单个核细胞中最相关的细胞类型是单核细胞和髓样树突状细胞(图 5b)。
- 这些发现与之前将单核细胞与 IBD 发病机制联系起来的研究一致。
Fig. 5: Elucidating GWAS traits through LINGER-inferred regulatory landscape.
- 图片说明- 经典单核细胞(上)和 CD56dim NK 细胞(下)中高表达转录因子和随机选择的转录因子的目标基因数量分布。使用 100 个最高表达的转录因子和 100 个随机选择的转录因子生成此分布。- IBD GWAS 在 PBMCs 细胞类型中的富集情况。气泡的颜色对应于高表达和随机选择的转录因子之间目标基因数量的比值比。x 轴是从单侧未配对 t 检验得到的高表达和随机选择的转录因子之间目标基因数量的 −log10(P 值)。- 经典单核细胞中关键的 IBD 相关调控因子。x 轴是所有转录因子中转录因子表达的 z 分数。y 轴是转录因子的调控分数。红色标记的转录因子是根据表达水平和调控分数之和排名最高的转录因子。- 经典单核细胞中 STAT1 靶基因中 IBD GWAS 基因的富集情况。小提琴图是通过随机选择 1,000 个转录因子生成的;STAT1 的重叠基因数量用星号标记。不同的小提琴图分别对应每个转录因子取前 200-5,000 个基因作为目标基因。- 经典单核细胞中 STAT1 靶基因中炎症活检与非炎症活检之间差异表达基因的富集情况。详情同 d。- 来自经典单核细胞转调节网络的 IBD 相关转录因子子网络。转录因子或目标基因节点的大小对应于其在网络中的度数。转录因子的颜色表示特征相关得分,目标基因的颜色表示分配给该基因的 GWAS SNP 的 −log10(P 值)。- SLC24A4 位点周围的顺式调控网络。相互作用表示显著的 RE–TG 链接,我们用启动子的位置来代表基因。
Para_03
- 我们接下来通过表达水平和性状调控评分的总和确定了关键转录因子。图5c列出了经典单核细胞中排名前八的候选转录因子。这些转录因子之前已被报道与炎症性肠病相关联。FOS可以增加炎症性肠病复发的风险;在炎症性肠病队列中鉴定出的一个变异位于ETV6的外显子上;IRF1和ETV6是具有活性差异的关键转录因子;基因FOS、FOSB和JUN编码炎症性肠病的强效介质;CUX1在炎症性肠病中被诱导表达;STAT1在表观遗传学上促进了炎症性肠病的发病机制。
Para_04
- 为了研究关键转录因子的下游靶标,我们选择了 STAT1 作为例子。
- 在经典单核细胞中由 STAT1 调控的前 200 个靶基因中,有 67 个与 GWAS 基因重叠,这一结果在基于随机选择的转录因子背景分布的情况下具有统计显著性(P 值小于 0.01,单侧自助法假设检验)。
- 对于前 500、1,000、2,000 和 5,000 个靶基因,重叠的数量也都具有显著性(图 5d)。
- 除了与 GWAS 相关的基因外,我们还收集了炎症活检和非炎症活检之间的差异表达基因,并发现这些差异表达基因与 STAT1 的顶级靶基因显著重叠(单侧自助法假设检验;图 5e)。
- 差异表达基因与 GWAS 基因之间没有显著重叠(P = 0.15,双侧 Fisher 精确检验),但差异表达基因和 GWAS 基因都与 STAT1 的顶级靶基因显著重叠,这表明了我们方法的稳健性和无偏性。
Para_05
- 最后,我们从经典的单核细胞转调控网络中提取了八个候选转录因子的子网络(图5f)。
- 我们还观察到,SLC24A4 的顺式调控网络(图5g)在与炎症性肠病相关的细胞类型中特别密集,该基因距离一个风险单核苷酸多态性 rs11626366 46 kb(P = 7.4 × 10^-3),这显示了不同细胞类型中疾病基因复杂的调控景观。
Identify driver regulators based on transcription profiles
基于转录谱识别驱动调节因子
Para_01
- 研究人员经常使用批量或单细胞表达数据来识别病例和对照之间的差异表达基因,但其潜在的调控驱动因素仍然不清楚。
- 转录因子活性,专注于转录因子蛋白质的DNA结合成分,是比mRNA更可靠的指标,用于识别驱动调控因子。
- 一种可行的方法是基于下游靶基因的表达模式来估计转录因子活性,这需要有一个准确的基因调控网络。
- 假设同一细胞类型在个体之间具有相同的基因调控网络结构,我们利用单个个体的单细胞多组学数据推断出的LINGER基因调控网络,仅使用相同细胞类型的基因表达数据来估计其他个体的转录因子活性。
- 通过比较病例和对照之间的转录因子活性,我们识别出了驱动调控因子。
- 这种方法非常有价值,因为它利用有限的单细胞多组学数据,仅使用基因表达数据来估计多个个体的转录因子活性(见方法)。
- 我们展示了两个示例,以说明其实用性。
Para_02
- 我们从26名急性髓系白血病(AML)患者和38名健康捐赠者中收集了批量基因表达数据。
- 我们基于从外周血单个核细胞(PBMCs)推断的细胞群体基因调控网络(GRN),计算了这些样本的转录因子活性,发现AML患者的FOXN1活性显著低于健康捐赠者,并且其表达没有差异(图6a,b)。
- 此外,我们还计算了671名AML个体的转录组谱(批量RNA-seq数据)的转录因子活性,并进行了生存分析,结果显示高FOXN1活性水平的个体往往具有更高的生存概率(图6c)。
- 此外,FOXN1已被报道为一种肿瘤抑制因子。
Fig. 6: Driver regulator identification.

- 图片说明- 健康供体(n = 38个独立样本)和AML患者(n = 26个独立样本)中FOXN1表达的小提琴图。均值表达没有显著差异(双侧非配对t检验)。- 健康供体(n = 38个独立样本)和AML患者(n = 26个独立样本)中FOXN1调控子活性的小提琴图(双侧非配对t检验,P = 0.035)。- 根据FOXN1调控子活性的AML生存分析(P值来自双侧对数秩检验)。- TCR刺激后0小时和8小时调控子活性和基因表达的热图。调控子活性差异的双侧非配对t检验,FOXK1和NR4A1的P值分别为0.0057和0.00081;基因表达的P值大于0.05。热图按行缩放。- TCR刺激后0小时、2小时、8小时和16小时全蛋白质组(wProtein)和磷酸化蛋白质组(pProtein)表达的热图。有两个生物学重复,分别表示为a和b。FOXK1和NR4A1的wProtein和pProtein表达在8小时时高于0小时。热图按行缩放。
Para_03
- 我们还提供了一个幼稚 CD4+ T 细胞在 T 细胞受体(TCR)刺激下的反应示例,这种刺激会诱导 T 细胞分化为各种效应细胞并激活 T 淋巴细胞。
- 我们基于幼稚 CD4+ T 细胞的基因调控网络(GRN)计算了转录因子(TF)活性,并识别了在 TCR 刺激后 8 小时与 0 小时相比差异活跃的调节因子。
- 根据调控子活性,FOXK2 和 NR4A1 在 8 小时时被激活(图 6d),这与整体蛋白质组学和磷酸蛋白质组学数据一致(图 6e)。
- 其他研究也表明 FOXK2 影响 T 淋巴细胞的激活,并揭示了 NR4A1 在调节性 T 细胞分化中的重要作用,这表明所鉴定的转录因子在 TCR 刺激下对幼稚 CD4+ T 细胞的反应中具有重要角色。
- 然而,FOXK2 和 NR4A1 在 8 小时时与 0 小时时相比表达没有显著差异(图 6d)。
In silico perturbation
计算机模拟扰动
Para_01
- 我们进行了计算机模拟扰动,以预测敲除转录因子后的基因表达。
- 为此,我们将单个转录因子或多个转录因子的表达量设置为零,并使用预测的基因表达作为计算机模拟扰动的基因表达。
- 我们利用计算机模拟扰动前后的表达差异来推断目标基因。
- 为了评估预测性能,我们从 KnockTF 数据库收集了血液细胞中八个转录因子的扰动数据作为真实值。
- 我们使用 LINGER 对这八个转录因子进行了计算机模拟个体扰动。
- 作为对比,我们还使用 CellOracle 和 SCENIC+ 方法进行了相同的计算扰动实验。
- 结果显示在扩展数据图 4c,d 中,表明 LINGER 比其他方法更准确(P ≤ 3.72 × 10−4)。
Para_02
- 为了评估 LINGER 推断分化行为的能力,我们使用 CellOracle 作为下游分析工具。
- 我们使用 LINGER 推断的基因调控网络(GRN)作为 CellOracle 的输入。
- 这使我们能够研究 LINGER 派生的网络再现分化反应的能力。
- 通过检查包含祖细胞群体的骨髓单核细胞数据,我们进行了 GATA1 的计算机模拟敲除实验,GATA1 是已知的红系和巨核细胞分化的重要调节因子。
- 基于 LINGER GRN 的 CellOracle 预测显示,GATA1 敲除将前体红细胞转移到了巨核细胞或红细胞祖细胞状态,这与 GATA1 在抑制红细胞成熟中的功能作用一致。
- 这些结果表明,LINGER 不仅可以预测基因表达在扰动下的变化,还可以通过与互补分析框架(如 CellOracle)的结合,实现对分化轨迹的下游表征。
Conclusions and discussions
Para_01
- LINGER 是一种基于神经网络的方法,通过结合批量数据集和转录因子-调控元件基序匹配知识,从配对的单细胞多组学数据中推断基因调控网络。
- 与现有工具相比,LINGER 实现了显著更高的基因调控网络推断准确性。
- 一个关键创新是终身机器学习,利用多样化的细胞环境,随着新数据的出现不断更新模型。
- 这解决了由于有限的单细胞数据集和庞大的参数空间阻碍复杂模型拟合的历史性挑战。
- LINGER 的终身学习方法具有在批量集合上预训练的优势,使用户可以轻松地为自己的研究重新训练模型,同时利用公共资源而无需直接访问这些资源。
- 传统上,基因调控网络推断性能是通过基因表达预测来评估的。
- 然而,使用终身学习利用外部数据并不会提高基因表达预测的准确性,但确实提高了基因调控网络的推断。
- 这一发现挑战了仅根据基因表达预测来评估基因调控网络推断的传统策略,并强调了考虑整体网络结构和调控相互作用的重要性。
Para_02
- 终身学习机制将鼓励模型在适应新的单细胞数据时保留来自大批量数据的先前知识。这是保留先前知识与拟合新数据之间的权衡。
- 在拟合新数据时,先前知识的变化灵活性不受限制。
- 最终结果偏离先前知识的程度取决于拟合新数据所产生的损失。
- LINGER 将自动学习这种权衡,以最大化利用两个数据集的信息。
Methods
GRN inference by lifelong learning
通过终身学习进行GRN推理
Para_01
- LINGER 是一个计算框架,用于从单细胞多组学数据中推断基因调控网络(GRNs)——即转录因子(TFs)、调控元件(REs)和靶基因(TGs)之间的成对调控关系。
- 总体而言,LINGER 通过基于神经网络模型的转录因子表达和调控元件染色质可及性来预测基因表达。
- 每个特征的贡献由神经网络模型的 Shapley 值估计,从而实现 GRNs 的推断。
- 为了捕捉大多数组织谱系的关键信息,LINGER 使用了终身机器学习(连续学习)。
- 此外,LINGER 通过在损失函数中引入流形正则化来整合基序结合数据。
Para_02
- LINGER 完整训练的输入数据包括外部批量和单细胞配对的基因表达和染色质可及性数据。
- 然而,我们提供了一个预训练的 LINGER 模型,该模型使用了批量数据,因此用户可以使用自己的单细胞数据重新训练该模型,而无需访问外部批量数据。
- 我们从 ENCODE 项目中收集了来自不同细胞环境的 201 个样本的配对批量数据——基因表达谱和染色质可及性矩阵。
- 单细胞数据是多组学单细胞数据的原始计数矩阵(RNA-seq 的基因计数和 ATAC-seq 的 RE 计数)。
- LINGER 使用神经网络架构为每个基因训练单独的模型,该架构包括一个输入层和两个全连接的隐藏层。
- 输入层的维度等于特征数量,包含待预测基因转录起始位点 (TSS) 前后 1 Mb 范围内的所有转录因子 (TFs) 和调控元件 (REs)。
- 第一个隐藏层有 64 个带有修正线性单元激活的神经元,可以捕捉由多个转录因子和调控元件组成的调控模块。
- 这些调控模块的特点是在相应调控元件上富集的转录因子基序。
- 第二个隐藏层有 16 个带有修正线性单元激活的神经元。
- 输出层是一个单一的神经元,它输出一个用于基因表达预测的实数值。 错误!!! - 待补充
Para_04
Para_08
- Fisher信息矩阵是基于训练于批量数据的神经网络计算得出的
GRN inference by Shapley value
基于Shapley值的GRN推断
Para_01
- Shapley值衡量了机器学习模型中特征的贡献,并广泛应用于深度学习、图形模型和强化学习等算法中。
- 我们使用样本间的绝对Shapley值的平均来推断转录因子(TF)和调控元件(RE)对目标基因(TGs)的调控强度,生成RE–TG顺式调控强度和TF–TG反式调控强度。
- 令({\beta }{{ij"}})表示调控元件j对目标基因i的顺式调控强度,({\gamma }{{ki"}})表示反式调控强度。
- 为了生成TF–RE结合强度,我们使用神经网络模型中输入层(TFs和REs)到第二层所有节点的权重来嵌入TF或RE。
- TF–RE结合强度是通过基于这种嵌入计算TF和RE之间的皮尔逊相关系数(PCC)得出的。
- ({\alpha }_{{kj"}})表示TF–RE结合强度。
Constructing cell type-specific GRNs
构建细胞类型特异的基因调控网络
Para_01
- 特定细胞类型的 TF–RE 调节潜力由以下公式给出:
Para_02
- 其中,TFBkj 是转录因子 k 和调控元件 j 之间的 TF–RE 调控潜力;sk 是转录因子 k 在细胞类型中的重要性得分,用于衡量转录因子激活细胞类型特异性开放染色质区域的偏好(将在下面的‘转录因子重要性得分’中描述);Ckj 是转录因子 k 和调控元件 j 之间的皮尔逊相关系数;Oj 是该细胞类型中所有细胞的平均染色质可及性;Bkj 是转录因子 k 和调控元件 j 之间的结合亲和力;αkj 是转录因子 k 和调控元件 j 之间的结合强度。
Para_03
- RE–TG 顺式调控潜能定义为:
Para_04
- 其中,CRPij 是转录因子 i 和调控元件 j 之间的顺式调控潜力;βij 是调控元件 j 对转录因子 i 的顺式调控强度;Oj 是平均染色质可及性;Ei 是细胞类型中所有细胞的平均基因表达;dij 是转录因子 i 和调控元件 j 之间基因组位置的距离;d0 是用于缩放距离的固定值,在本文中设置为 25,000。
Para_05
- TF–TG 跨调控潜能被定义为相应反应元件对 TG 的累积效应:
Para_06
- 其中,γ_ki 是转录因子 k 和目标基因 i 之间的 TF–TG 转录调控强度;Si 是目标基因 i 的转录起始位点(TSS)前后 1 Mb 范围内的调控元件集合;CRP_ij 是目标基因 i 和调控元件 j 之间的顺式调控潜力;TFB_kj 是转录因子 k 和调控元件 j 之间的 TF–RE 调控潜力。
Constructing cell-level GRNs
构建细胞水平的基因调控网络
错误!!! - 待补充
TF importance score
TF重要性分数
Para_01
- 为了系统地识别在控制细胞类型染色质可及性方面发挥关键作用的转录因子,我们引入了一个转录因子重要性评分。
- 该评分旨在衡量转录因子激活细胞类型特异性调控元件的偏好程度。
- 输入数据是带有已知细胞类型注释的多组学单细胞数据。
- 生成转录因子重要性评分分为四个步骤:
TF–RE binding affinity matrix
TF–RE结合亲和力矩阵
Para_01
- 我们从PECA284的GitHub页面下载了713个已知motif的转录因子位置权重矩阵,这些数据收集自广泛使用的数据库,包括JASPAR、TRANSFAC、UniPROBE和Taipale。
- 给定一组调控元件,我们使用Homer进行motif扫描,计算每个转录因子的结合亲和力得分,作为衡量转录因子与调控元件之间相互作用强度的定量指标。
Identify motif-binding REs
识别基序结合的调控元件
Para_01
- 我们通过使用 Homer86 进行基序扫描来识别与基序结合的 REs。
ChIP–seq-based validation
基于 ChIP–seq 的验证
Para_01
- 鉴于选择的转录因子可能会影响最终结果,我们使用以下标准从 Cistrome 数据库中收集所有满足以下条件的 ChIP–seq 数据。
Para_02
- 选择PBMC的ChIP–seq数据的程序如下。 [ul]- We downloaded all human TF ChIP–seq information, including 11,349 datasets. - We filtered samples that did not pass quality control, and 4,657 datasets remained. - We chose samples in blood tissue, and 609 datasets remained. - We filtered the cell line data that is not consistent with PBMC cell types, and 63 datasets remained. - We chose the TF expressed in single-cell data and with known motifs available, and 39 datasets remained. - We chose the experiments that were done in one of the 14 cell types detected in the PBMC data, and 20 datasets remained.
Para_03
- 选择 H1 细胞系的 ChIP–seq 数据的程序如下: [ul]- We downloaded all human TF ChIP–seq information, including 11,349 datasets. - We filtered samples that did not pass quality control, and 4,657 datasets remained. - We chose the H1 cell line, and 42 datasets remained. - We chose the TF expressed in single-cell data and with known motifs available, and 33 datasets remained.
Perturbation-based validation
基于扰动的验证
Para_01
- 从 KnockTF 数据库中选择真实数据的标准类似于 ChIP–seq 数据。
Para_02
- 选择PBMC敲低数据的程序如下。 [ul]- We selected the molecular type as ‘TF’ and chose the ‘Peripheral_blood’ tissue type, with 21 cases remaining. - There are 11 datasets included in the PBMCs cell type in the single-cell data. - We chose the TF expressed in single-cell data and with known motifs available, and 8 datasets remained.
PBMC 10× data
PBMC 10× 数据
Para_01
- 我们从 10× Genomics 网站 (https://support.10xgenomics.com/single-cell-multiome-atac-gex/datasets) 下载了 PBMC 10K 数据。注意,该数据集包含 11,909 个细胞,并且通过细胞分选去除了粒细胞。
- 我们使用 10× Genomics 软件 Cell Ranger ARC 输出的特征矩阵过滤后的细胞作为输入,并进行下游分析。
- 首先,我们在 Seurat(版本 4.0)中执行加权最近邻分析,移除了 1,497 个细胞。
- 我们还移除了没有替代真实标签的细胞,最终得到 9,543 个细胞。
- 我们通过随机选择每种细胞类型的细胞数的平方根,并对最近的 100 个细胞的表达水平和染色质可及性进行平均,生成了元细胞数据,以产生选定细胞的基因表达和染色质可及性值。
- 元细胞数据直接输入 LINGER 进行分析。
AUPR ratio
AUPR比率
Para_01
- 为了衡量预测器的准确性,我们定义了AUPR比率,即一种方法的AUPR与随机预测器的AUPR之比。
- 对于随机预测器,AUPR等于数据集中正样本的比例。
- AUPR比率定义为预测器相对于随机预测的准确性的倍数变化,表示为正样本数量与实际正样本数量的比值。
Data availability
Para_01
- 本研究中使用的PBMC数据是从10× Genomics网站(https://s3-us-west-2.amazonaws.com/10x.files/samples/cell-arc/1.0.0/pbmc_granulocyte_sorted_10k/pbmc_granulocyte_sorted_10k_fastqs.tar)下载的。
- SNARE-seq数据从NCBI基因表达数据库(https://www.ncbi.nlm.nih.gov/geo/)下载,访问编号为GSE126074。
Code availability
Para_01
- 该软件可在 GitHub (https://github.com/Durenlab/LINGER) 和 Zenodo 仓库中获取,采用 GPLv3 许可证。
- 我们在这项研究中使用了 Python 和 R。
腾讯云开发者
