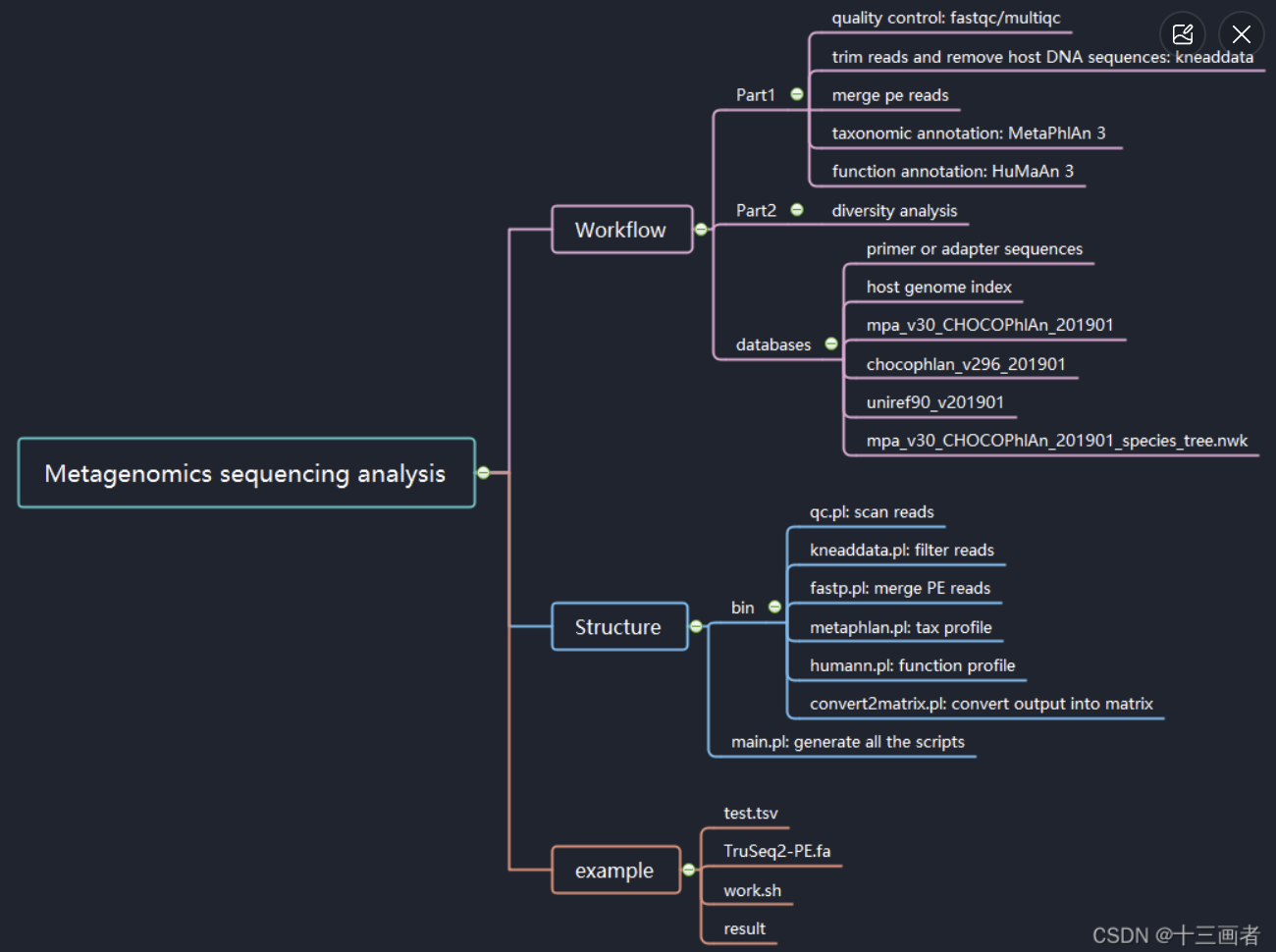
提取信息
关注我,不错过每一次更新。

#!/usr/bin/perl -w
open IN,"ARGV[0]" or die $!;
open OUT,">ARGV[1]" or die $!;
while(<IN>){
chomp;
my @arr=split /\t/;
if(($_=~/^#/)||($arr[]=~/^INDEL.*/)||($arr[] =~ /,/)||($arr[] < 20)){ ###提取信息
next;
}
else{
print OUT $_,"\n";
}
}
close IN;
close OUT;
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2016-04-16,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
暂无评论
推荐阅读
编辑精选文章
换一批









推荐阅读
相关推荐
简单实例-对比文件
更多 >
腾讯云开发者

